En automatisk procedure, der er skrevet i Microsoft Visual Basic (VB), 6.,0 opdateringer DAVID ugentligt med følgende procedurer: ring til en serie af Perl og Java-applikationer, der kan downloades offentlige data via anonym file transfer protokol (FTP) (Tabel 1); pak og parse ønskede kommentering af data, oprette tabulatorsepareret data filer klar til database indførsel og import af data til en Oracle 8i relationel database management system (RDBMS) ved hjælp af Oracle”s SQL*Loader. Microsoft”s IIE web server og Active Server Page teknologi, der bruges til at få adgang til databasen ved hjælp af JavaBeans og structured query language (SQL)., LocusLink tal for Affymetrix probe sæt er afledt fra University of Michigan foreninger eller NetAffx . Funktionelle anmærkninger og database krydshenvisninger er afledt af LocusLink, som giver stabile, human-kuraterede repræsentationer af gener. For mere detaljerede oplysninger om de datakilder, der anvendes af DAVID se FA .sektion på.,
Analyse moduler
DAVID er sammensat af fire moduler: Annotation Tool, GoCharts, KeggCharts, og DomainCharts. Annotationsværktøjet er en automatiseret metode til funktionel annotation af genlister. Enhver kombination af annotationsdata kan vælges mellem 10 indstillinger ved at vælge de relevante afkrydsningsfelter (tabel 2)., De anmærkninger føjes til den indsendte gen liste ved at vælge knappen upload, som returnerer en HTML-tabel, der indeholder brugerens oprindelige liste over identifikatorer vedlagt de valgte funktionelle anmærkninger. Unannoterede gener er inkluderet i output uden vedhæftede data til sporingsformål.,
GoCharts modul grafisk viser fordelingen af differentielt udtrykte gener blandt funktionelle kategorier ved hjælp af kontrollerede ordforråd af Genet Ontologi Consortium (GO), som giver et struktureret sprog, der kan anvendes til de funktioner, gener og proteiner i alle organismer selv som viden fortsætter med at akkumulere og forandring ., Sproget er struktureret i en rettet acyklisk graf (DAG), hvor term specificitet stiger og genom dækning falder som man bevæger sig ned i hierarkiet. I modsætning til en sand hierarki, barn vilkår i en DAG kan have mere end parentn forælder sigt og kan have en anden klasse af forhold til sine forskellige forældre. Strukturen af GO starter med tre hovedkategorier, biologisk proces, Molekylær funktion og cellulær komponent., Biologisk proces omfatter brede biologiske mål, såsom mitose eller purinmetabolisme, der opnås ved ordnede samlinger af molekylære funktioner. Molekylær funktion beskriver de opgaver, der udføres af individuelle genprodukter; eksempler er transkriptionsfaktor og DNA-helikase. Den cellulære komponent klassifikation type involverer subcellulære strukturer, steder, og makromolekylære komplekser; eksempler indbefatter kerne, telomere, og oprindelse anerkendelse kompleks., Når du har valgt en klassifikationstype, vælges niveauer, der bestemmer listedækning og specificitet, ved at vælge den relevante radioknap. Niveau 1 giver den højeste listedækning med den mindste mængde term specificitet. Med hvert stigende niveau falder dækningen, mens specificiteten stiger, så niveau 5 giver den mindste mængde dækning med den højeste term specificitet.
klassificeringsdata vises som et søjlediagram, hvor længden af linjen repræsenterer antallet af genidentifikatorer i hver kategori., Brugeren kan indstille visualiseringsparametre til sortering af outputdata og visning af kategorier, der indeholder mindst et minimum antal gener. Valg af en individuel bjælke åbner en ny HTML-tabel, der viser genidentifikatoren, LocusLink-nummeret, gennavnet, den aktuelle klassificering og andre klassifikationer for hvert gen i den kategori. En” vis alle ” – knap åbner en ny HTML-tabel, der viser alle klassificeringsdata, og en “vis diagramdata” – knap åbner en HTML-tabel, der indeholder de underliggende diagramdata, hvilket giver brugerne mulighed for at genskabe tilpasset diagramgrafik i et regnearksprogram., Et nyt diagram kan blive vist for enhver delmængde af gener ved at vælge klassificering type og niveau ved hjælp af afkrydsningsfelter og radioknapper til rådighed, inden brugeren”s aktuelle side, der giver mulighed for drill-down funktionalitet. Et antal af antallet af gener, der er kommenteret, er inkluderet i output, og ikke-annoterede gener er binned i kategorien “uklassificeret”, hvilket giver brugerne et automatiseret sporingssystem for gener, der ikke er kommenteret.
KeggCharts viser grafisk fordelingen af differentielt udtrykte gener blandt Kegg biokemiske veje., Hver vej er knyttet til Kegg-Path pathwayay-kortet, hvor differentielt udtrykte gener fra den oprindelige liste fremhæves med rødt. I denne opfattelse gener er yderligere knyttet til yderligere anmærkninger tilgængelige via KEGG ” s DBGET søgesystem . Som med GoCharts, kan brugeren indstille visualisering parametre til sortering af output data og visning af kategorier, der indeholder mindst et minimum af gener og KeggCharts visualisering arver alle de dynamiske funktioner i GoCharts.
DomainCharts viser fordelingen af differentielt udtrykte gener blandt PFAM-proteindomæner ., Hver domænebetegnelse er knyttet til Conserved Domain Database (CDD) fra National Center for Biotechnology Information (NCBI), hvor detaljer vedrørende domænefunktion, struktur og sekvens er let tilgængelige. Som med GoCharts og KeggCharts, kan brugeren indstille visualisering parametre til sortering af output data og visning af kategorier, der indeholder mindst et minimum af gener og DomainCharts visualisering arver alle de dynamiske funktioner i GoCharts og KeggCharts. For yderligere information om funktionaliteten af DAVID besøge fa .sektion på.,
Bruger DAVID til mine funktionelle annotation
for At demonstrere funktionaliteten af DAVID analyserede vi en liste af gener differentielt udtrykte i humant perifert blod mononukleære celler (PBMCs) efter inkubation med HIV-1 kuvert proteiner. Nærmere oplysninger om de eksperimentelle, RNA forberedelse, og GeneChip hybridisering procedurer, sammen med detaljer af chip-til-chip normaliseringer og statistisk analyse af differential genekspression er tilvejebragt i Cicala et al. ., Kort fortalt blev primære humane Pbmc ‘ er og monocytafledte makrofager inkuberet i 16 timer med HIV-1 kuvertprotein (gp120). Oligonukleotidmikroarrays med høj densitet (Affymetri.HU-95A GeneChip) blev brugt til at overvåge gp120-inducerede transkriptionelle begivenheder. Denne analyse resulterede i identifikation af 402 differentielt udtrykte gener.
der Henviser til, at 16 gener moduleret af HIV-1 gp120 har tidligere været forbundet med en HIV-smittet og/eller konvolut, signalering, de resterende gener er af ukendt funktion eller har aldrig været forbundet med HIV-1 eller gp120., Konvertering af denne liste over gener til biologisk betydning kræver indsamling af relevant information fra flere datalagre. Mange forskere mener at denne proces består af iterative browsing gennem flere databaser for hvert gen, manuelt indsamle gen-specifikke oplysninger om, sekvens, funktion, gangsti, og disease association. I modsætning hertil tilføjer Davids systematiske tilgang samtidig biologisk rig information afledt af flere offentlige datakilder til lister over gener parallelt., Valg DAVID ” s Annotation værktøj og uploade listen over 402 differentielt udtrykte gener initierer den funktionelle annotation og analyse af hele datasættet. Når den er indsendt, gemmes genlisten for hele analysesessionen, så brugerne kan skifte mellem moduler uden at skulle indsende data igen.
annotationsværktøj
Annotationsværktøjet indeholder flere annotationsindstillinger og bygger en tabelvisning af brugernes genliste og de tilgængelige annotationer (tabel 2)., At vælge annotationen felter Gene Symbol, LocusLink, OMIM, Unigene, Reference Sekvens, og Gen Navn, der blev fulgt på ved at vælge “Upload” – knappen producerer en HTML-tabel i web browser, der indeholder alle gener og deres rådighed annotationer, hvor gen-id, beskrivende og klassificering af data er trukket fra databasen og er knyttet til gene liste (Figur 1). Genidentifikatorer såsom Gensymbol og LocusLink er hyperlinket til yderligere genspecifikke data, der er tilgængelige på deres oprindelige kilder, hvilket giver dybdegående genspecifikke detaljer og annotation stamtavler., Klassifikation data og funktionelle resum .er kan bruges til hurtigt at scanne efter oplysninger, der er relevante for forskeren”s eksperimentelle system. Serveren tid, der kræves til udførelse af dette modul korrelerer lineært med størrelsen af genet listen, og tager mindre end 45 sekunder til listerne på op til 1.000 gener (Figur 2, tal i parentes repræsenterer r2 værdier). Disse resultater viser styrken og effektiviteten af en integreret tilgang til den funktionelle annotation af store datasæt.,

Udsend af Annotation Tool. Vist er vedlagt anmærkninger for de første flere Affymetri.probe sæt i en HTML-tabel, der indeholder alle 402 poster. Kategoriske oplysninger om forsøgsbetingelserne blev indsendt sammen med Affymetri.probesætidentifikatorer og inkluderet i output i værdikolonnen. Identifikatorer som Symbol, LocusLink, omim, Refse.og Unigene accessions er hyperlinket til deres oprindelseskilder for mere detaljerede oplysninger., Tekst indgår i sammenfattende felter er afledt af beskrivende, funktionelle oplysninger i NCBI ” s LocusLink rapporter.
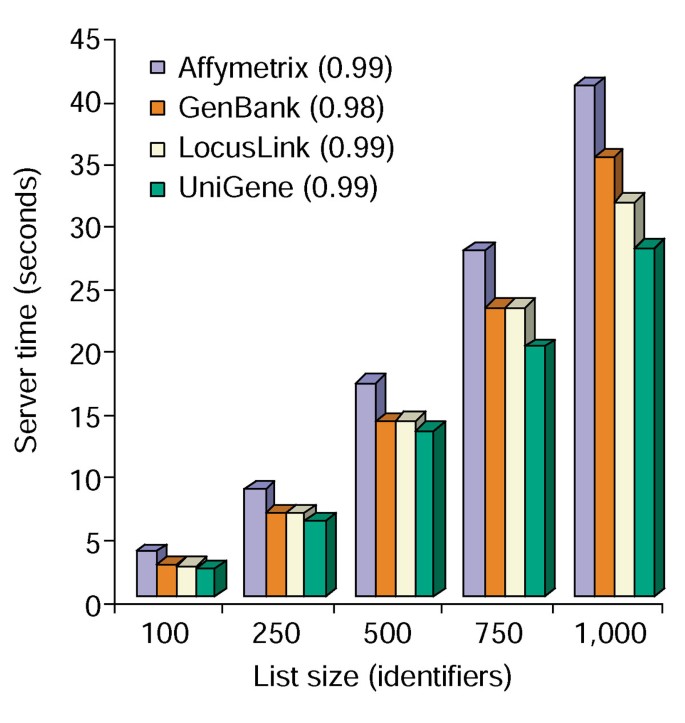
Tiden analyse af Annotation Tool. Server tid, der kræves (Y-akse) for samtidig at tilføje alle 10 annotation muligheder til Gen lister spænder i størrelse fra 100 til 1.000 (axis-akse)., Gennemsnittet af tre forsøg for genlister, der indeholder Affymetri., GenBank, LocusLink og UniGene identifikatorer, vises, og tallene i parentes repræsenterer R2-værdien af korrelationen mellem gen-listestørrelse og den Servertid, der kræves til annotation.
GoCharts
valg af GoCharts-modulet åbner et nyt vindue med forskellige muligheder., Brugere vælger mellem tre generelle typer klassificering (biologisk proces, molekylær funktion og cellulær komponent) og fem niveauer af annotation, der repræsenterer term dækning og specificitet (se Analysemoduler afsnit). Enhver kombination af klassificering og dækningsniveau kan specificeres. Også inkluderet er muligheder for at anmærke gen lister med alle GO vilkår tilgængelige eller kun de mest specifikke vilkår, der omtales som terminal noder., Muligheden for at vælge forskellige niveauer af term specificitet giver den nødvendige fleksibilitet og giver således forskere mulighed for dynamisk at bestemme hvilket niveau af dækning og specificitet der bedst passer til deres data og analysefase. For eksempel kan analyser på et tidligt stadium bestå i at annotere genlister med meget generelle termer for at opnå en bred forståelse af dataene. I dette tilfælde klassificerer valg af biologisk proces og niveau 1 gener ved hjælp af generelle udtryk som “død” og “cellekommunikation”., Brug af øget term specificitet Letter udvindingen af mere detaljerede funktionelle oplysninger. I dette tilfælde vælger biologisk proces og niveau 5 klassificerer gener ved hjælp af udtryk som “apoptotiske mitokondrielle ændringer” og “kemosensorisk opfattelse”.
men øget sigt specificitet kommer en omkostning, idet det øger listen dækning falder (figur 3). I vores undersøgelser finder vi, at Niveau 2 typisk opretholder god dækning og samtidig giver meningsfuld term specificitet., Figur 4a illustrerer, hvordan GoCharts-visualiseringen hurtigt afslører, at 35 differentielt udtrykte gener er involveret i”stressresponser”. Hver go sigt kan ses i træet eller DAG visninger af hyperlinks til Quickuickgo .
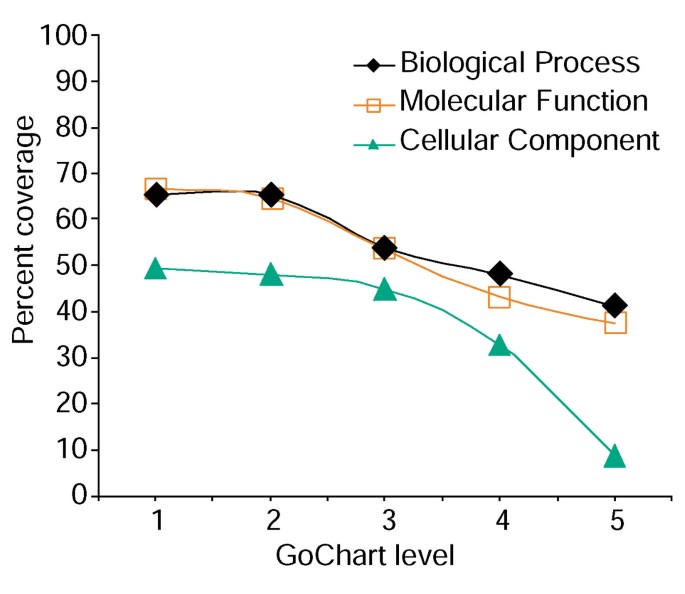
Analyse af gen-liste dækning ved hjælp af GoCharts. En liste over 402 Affymetri.sonde sæt identifikatorer blev kommenteret med Proteome tildelt funktionelle klassifikationer leveret af LocusLink., Procent dækning repræsenterer antallet af gener ud af 402, der blev kommenteret på et term-specificitetsniveau inden for den biologiske proces, Molekylær funktion og cellulære Komponentklassifikationstyper. Procent dækning falder som sigt specificitet stiger.
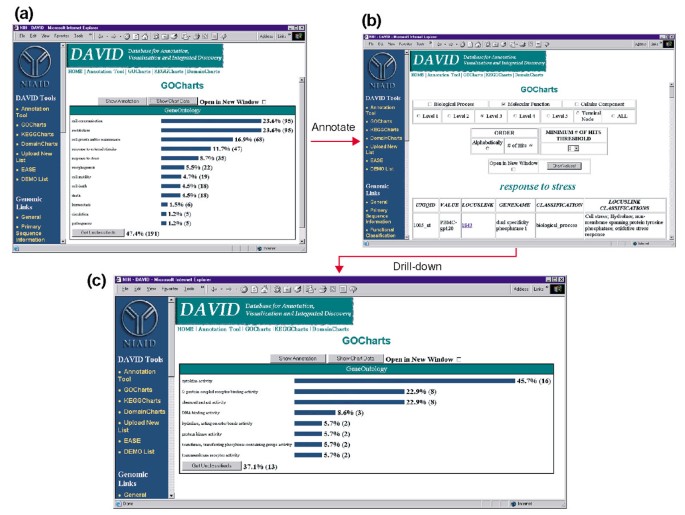
Udsend af GoCharts. (a) et søjlediagram, der viser fordelingen af differentielt udtrykte gener blandt Gen ontologi (GO) biologiske processer., Parametre blev sat til at gå niveau 2, et hit tærskel på fem, og output blev sorteret efter hit tæller. Blå bjælker er knyttet til yderligere annotationsdata vist i (b). Valg af den blå bjælke i (a) svarende til “respons på stress” åbner en HTML-tabel, der viser LocusLink, gennavn, nuværende klassificering og andre klassificeringsdata for generne i den kategori. (c) denne undergruppe af gener involveret i “stressrespons” blev yderligere karakteriseret ved at vælge GO-Molekylær funktion, GO-niveau 3, En hit-tærskel på 2 og sorteret efter hitantal., Valg af knappen “Diagramværdier” opretter et nyt histogram, der afslører, at 16 af de 35 stress-Responsgener koder for proteiner, der besidder cytokinaktivitet.,
på Grund af HIV-1 har en stor indflydelse på funktionen af celler i immunsystemet og deres evne til at udføre stress-reaktioner, vi har valgt histogram bar, der repræsenterer antallet af gener involveret i stress-respons, som åbner en HTML-tabel, der indeholder de Affymetrix identifikator, LocusLink antal, gen-navn, den nuværende klassificering, og andre klassifikationer for alle gener 35 (Figur 4b)., Nu hvor vi har reduceret vores genliste til de gener, der er involveret i stressresponser, karakteriserede vi yderligere denne delmængde ved at gentage GoCharts-proceduren, der er tilgængelig øverst i stress-respons HTML-tabellen. Ved at vælge molekylær funktion producerer niveau 3 et nyt histogram, der hurtigt afslører, at næsten halvdelen (16/35) af stress-responsgenerne har cytokinaktivitet (figur 4c)., Faktisk har cytokiner vist sig at spille en vigtig rolle i HIV-1 livscyklus, og de opnåede resultater antyder, at behandling af Pbmc ‘ er med HIV-1 kuvertproteiner signifikant modulerer transkriptionen af adskillige cytokingener. Den effektivitet, hvormed GoCharts systematisk sammenfattet dette store datasæt med grafiske visualiseringer, mens de resterende knyttet til primære data og eksterne ressourcer drastisk forbedret discovery-processen.,
KeggCharts
Figur 5a viser produktionen af KeggCharts med et histogram, der viser fordelingen af differentielt udtrykte gener blandt biokemiske pathways. Diagrammet viser, at en KEGG-vej til apoptose inkluderer fem gener induceret af HIV-1 gp120. At vælge den vej, navn, åbnes den tilsvarende KEGG biokemisk pathway kort og fremhæver i rødt omrids differentielt udtrykte gener fungerer i, at vejen (Figur 5b). I denne opfattelse gener er yderligere knyttet til yderligere anmærkninger tilgængelige via KEGG ” s DBGET søgesystem ., Bemærk, at kun fire gener i Kegg-apoptosevejen fremhæves med rødt, mens keggcharts-værktøjet kortlagde fem Affymetri. – sonde sæt til apoptosevejen. Denne forskel skyldes det faktum, at to af Affymetri.-probesetene er rettet mod det samme “TNF-alpha” – gen.
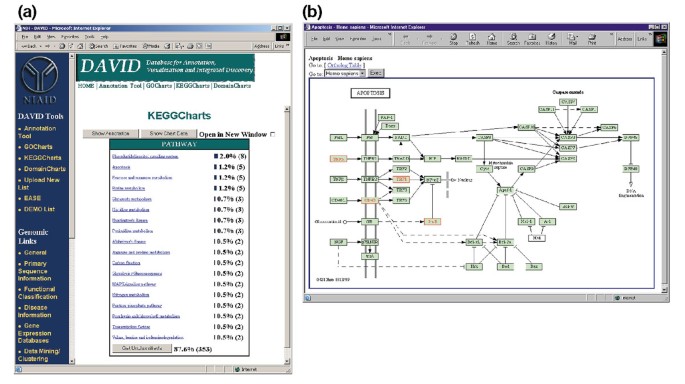
Udsend af KeggCharts. (a) Visualiseringsdiagram, der viser fordelingen af 402 gener mellem Kegg-biokemiske veje. Den ramte tærskel blev sat til tre og output blev sorteret efter hit tæller., Det store antal uklassificerede identifikatorer skyldes, at KEGG er biokemisk-vejcentrisk og således giver lav dækning af genlister. På samme måde som output af GoCharts repræsenterer blå søjler antallet af gener i hver vej. Valg af en blå bjælke åbner en HTML-tabel, der viser LocusLink, gennavn, nuværende klassificering og andre klassificeringsdata for generne i den vej (data ikke vist)., (b) den biokemiske Kegg-vej, der vises efter udvælgelsen af vejnavnet “apoptose” i (a), viser fire differentielt udtrykte gener inden for apoptosevejen ved at fremhæve dem i lysegrøn og rød. Det faktum, at KEGG-vejen kun fremhæver fire gener, mens KeggChart kortlægger fem Affymetri. – probesæt til apoptosevejen skyldes det faktum, at to probesæt målretter mod det samme “TNF-alpha” – gen.,
DomainCharts
DomainCharts er driftsmæssigt beslægtet med både KeggCharts og GoCharts, bortset fra, at de resultater, der visuelt afbilder fordelingen af gener blandt PFAM protein-domæner (Figur 6a). DomainCharts-histogrammet identificerer 16 gener med kinasedomæner (pkinase), hvilket sandsynligvis afspejler virkningerne af HIV-1 gp120 på signaltransduktionsmaskineriet. Diagrammet identificerer også seks gener med interleukin-8-domæner (IL-8), et domæne, der repræsenterer et stærkt bevaret motiv blandt stressresponscytokiner., Hvis du vælger domænenavnet “IL8”, åbnes siden Conserved Domain Database (CDD) svarende til PFAM-domænet (figur 6b). Denne side indeholder detaljeret sekvens, struktur og funktionelle oplysninger om IL-8-domænet og de proteiner, der indeholder det.
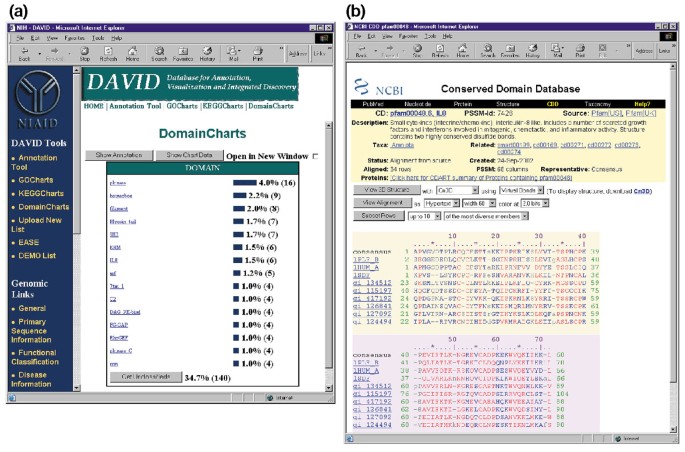
Udsend af DomainCharts. (a) Visualiseringsdiagram, der viser fordelingen af 402 gener blandt proteindomæner. Parametrene blev sat til et minimum hit tærskel på fire og output blev sorteret efter hit tæller., I lighed med output fra GoCharts og KeggCharts repræsenterer blå søjler antallet af gener, der indeholder det pågældende domæne. Valg af en blå bjælke åbner en HTML-tabel, der viser LocusLink, gennavn, nuværende klassificering og andre klassificeringsdata for generne i den vej (data ikke vist)., b) Valg af domæne navn “IL8” i (a), som indeholder seks differentielt udtrykte gener, bringer brugeren til en ny side, der indeholder outputtet fra de Bevarede Domæne Database (CDD) af NCBI, som indeholder detaljerede oplysninger om IL-8 domæne, herunder strukturelle information, flere sekvens linjeføringer, samt beskrivende oplysninger om domænet og de proteiner, der besidder det.