une procédure automatisée écrite dans Microsoft Visual Basic (VB) 6.,0 met à jour DAVID chaque semaine avec les procédures suivantes: appeler une série D’applications Perl et Java qui téléchargent des données publiques via des protocoles de transfert de fichiers anonymes (FTP) (Tableau 1); décompresser et analyser les données d’annotation souhaitées; créer des fichiers de données délimités par des onglets prêts pour l’importation de base de données; et importer des Le serveur web IIe de Microsoft et la technologie Active Server Page sont utilisés pour accéder à la base de données en utilisant JavaBeans et le langage de requête structuré (SQL)., Les numéros LocusLink pour les ensembles de sondes Affymetrix sont dérivés d’associations de L’Université du Michigan ou de NetAffx . Les annotations fonctionnelles et les références croisées de base de données sont dérivées de LocusLink, qui fournit des représentations stables de gènes organisées par l’homme. Pour plus d’informations sur les sources de données utilisées par DAVID, veuillez consulter la section FAQ À l’adresse suivante:.,
l’Analyse des modules
DAVID est composé de quatre modules principaux: Outil d’Annotation, GoCharts, KeggCharts, et DomainCharts. L’Outil d’Annotation est une méthode automatisée pour l’annotation fonctionnelle des listes de gènes. Toute combinaison de données d’annotation peut être choisie parmi 10 options en cochant les cases appropriées (Tableau 2)., Les annotations sont ajoutées à la liste de gènes soumis en sélectionnant le bouton de téléchargement, qui renvoie un tableau HTML contenant la liste originale de l »Utilisateur des identifiants ajoutés avec les annotations fonctionnelles choisies. Les gènes non annotés sont inclus dans la sortie sans données ajoutées à des fins de suivi.,
le module GoCharts affiche graphiquement la distribution des gènes exprimés de manière différentielle entre les catégories fonctionnelles en utilisant le vocabulaire contrôlé du Gene Ontology Consortium (GO), qui fournit un langage structuré fonctions des gènes et des protéines dans tous les organismes alors même que les connaissances continuent de s’accumuler et de changer ., Le langage est structuré dans un graphe acyclique dirigé (DAG), dans lequel la spécificité du terme augmente et la couverture du génome diminue à mesure que l’on descend dans la hiérarchie. Contrairement à une véritable hiérarchie, les Termes enfants dans un DAG peuvent avoir plus d’un terme parent et peuvent avoir une classe de relation différente avec ses différents parents. La structure de GO commence par trois catégories principales, le processus biologique, la fonction moléculaire et la composante cellulaire., Le processus biologique comprend de larges objectifs biologiques, tels que la mitose ou le métabolisme des purines, qui sont accomplis par des assemblages ordonnés de fonctions moléculaires. La fonction moléculaire décrit les tâches effectuées par des produits génétiques individuels; des exemples sont le facteur de transcription et L’ADN hélicase. Le type de classification des composants cellulaires implique des structures subcellulaires, des emplacements et des complexes macromoléculaires; des exemples incluent le noyau, le télomère et le complexe de reconnaissance d’origine., Après avoir choisi un type de classification, les niveaux qui déterminent la couverture et la spécificité de la liste sont choisis en sélectionnant le bouton radio approprié. Le niveau 1 fournit la couverture de liste la plus élevée avec le moins de spécificité de terme. Avec chaque niveau croissant, la couverture diminue tandis que la spécificité augmente, de sorte que le niveau 5 fournit le moins de couverture avec la spécificité à terme la plus élevée.
Les données de Classification sont affichées sous forme de diagramme à barres, où la longueur de la barre représente le nombre d’identificateurs de gènes dans chaque catégorie., L’utilisateur peut définir des paramètres de visualisation pour trier les données de sortie et afficher des catégories contenant au moins un nombre minimum de gènes. La sélection d’une barre individuelle ouvre un nouveau tableau HTML affichant l’identifiant du gène, le numéro de LocusLink, le nom du gène, la classification actuelle et d’autres classifications pour chaque gène de cette catégorie. Un bouton » Afficher tout « ouvre un nouveau tableau HTML affichant toutes les données de classification et un bouton » Afficher les données de graphique » ouvre un tableau HTML contenant les données de graphique sous-jacentes, permettant ainsi aux utilisateurs de recréer des graphiques de graphique personnalisés dans un tableur., Un nouveau graphique peut être affiché pour n »importe quel sous-ensemble de gènes en sélectionnant le type et le niveau de classification en utilisant les cases à cocher et les boutons radio disponibles dans la page actuelle de l » utilisateur qui permettent des capacités d » exploration. Un décompte du nombre de gènes annotés est inclus dans la sortie, et les gènes non annotés sont regroupés dans la catégorie « non classifié », fournissant ainsi aux utilisateurs un système de suivi automatisé pour les gènes non annotés.
Les diagrammes de Kegg montrent graphiquement la distribution des gènes exprimés différentiellement entre les voies biochimiques de KEGG., Chaque voie est liée à la carte de voie de KEGG, dans laquelle les gènes exprimés différentiellement de la liste originale sont surlignés en rouge. Dans cette vue, les gènes sont en outre liés à des annotations supplémentaires disponibles via le système de récupération DBGET de KEGG . Comme avec GoCharts, l’utilisateur peut définir des paramètres de visualisation pour trier les données de sortie et afficher des catégories contenant au moins un nombre minimum de gènes et la visualisation KeggCharts hérite de toutes les fonctionnalités dynamiques de GoCharts.
Les diagrammes de domaines montrent la distribution des gènes différentiellement exprimés entre les domaines protéiques PFAM ., Chaque désignation de domaine est liée à la base de données des domaines conservés (CDD) du National Center for Biotechnology Information (NCBI), où des détails concernant la fonction, la structure et la séquence du domaine sont facilement disponibles. Comme avec GoCharts et KeggCharts, l’utilisateur peut définir des paramètres de visualisation pour trier les données de sortie et afficher des catégories contenant au moins un nombre minimum de gènes et la visualisation DomainCharts hérite de toutes les fonctionnalités dynamiques de GoCharts et KeggCharts. Pour plus d’informations sur la fonctionnalité de DAVID, visitez la section FAQ À .,
utiliser DAVID pour exploiter l’annotation fonctionnelle
pour démontrer la fonctionnalité de DAVID, nous avons analysé une liste de gènes exprimés de manière différentielle dans les cellules mononucléées du sang périphérique humain (PBMC) après incubation avec des protéines D’enveloppe du VIH-1. Des détails sur les procédures expérimentales, de préparation D’ARN et D’hybridation GeneChip, ainsi que des détails sur les normalisations de puce à puce et l’analyse statistique de l’expression différentielle des gènes sont fournis dans Cicala et al. ., En bref, les PBMC primaires humaines et les macrophages dérivés de monocytes ont été incubés pendant 16 heures avec la protéine D’enveloppe du VIH-1 (gp120). Des puces à oligonucléotides de haute densité (Affymetrix hu-95a GeneChip) ont été utilisées pour surveiller les événements transcriptionnels induits par le gp120. Cette analyse a permis d’identifier 402 gènes d’expression différentielle.
alors que 16 gènes modulés par le VIH-1 gp120 ont déjà été associés à la réplication du VIH et / ou à la signalisation de l’enveloppe, les gènes restants ont une fonction inconnue ou n’ont jamais été associés au VIH-1 ou au gp120., La conversion de cette liste de gènes en signification biologique nécessite la collecte d’informations pertinentes provenant de plusieurs dépôts de données. Pour de nombreux chercheurs, ce processus consiste à parcourir plusieurs bases de données itératives pour chaque gène, en collectant manuellement des informations spécifiques au gène concernant la séquence, la fonction, la voie et l’association des maladies. En revanche, L’approche systématique de DAVID ajoute simultanément des informations biologiquement riches dérivées de plusieurs sources de données publiques à des listes de gènes en parallèle., La sélection de L’outil D’Annotation de DAVID et le téléchargement de la liste des 402 gènes exprimés de manière différentielle initient l’annotation fonctionnelle et l’analyse de l’ensemble de données. Une fois soumise, la liste de gènes est stockée pendant toute la session d’analyse, ce qui permet aux utilisateurs de basculer entre les modules sans avoir à soumettre de nouveau des données.
outil D’Annotation
l’outil D’Annotation fournit plusieurs options d’annotation et crée une vue tabulaire de la liste des gènes des utilisateurs et des annotations disponibles (Tableau 2)., Le choix des champs d’annotation Gene Symbol, LocusLink, OMIM, Unigene, Reference Sequence et Gene Name suivi de la sélection du bouton « Upload » produit un tableau HTML dans le navigateur Web contenant tous les gènes et leurs annotations disponibles, où les identifiants de gènes, les données descriptives et de classification sont extraits de la base de données et ajoutés à la liste des gènes (Figure 1). Les identificateurs de gènes tels que Gene Symbol et LocusLink sont hyperliés à des données spécifiques de gènes supplémentaires disponibles à leurs sources d’origine, fournissant ainsi des détails spécifiques de gènes en profondeur et des pedigrees d’annotation., Les données de Classification et les résumés fonctionnels peuvent être utilisés pour rechercher rapidement des informations pertinentes pour le système expérimental du chercheur. Le temps de serveur requis pour l’exécution de ce module est corrélé linéairement à la taille de la liste de gènes et prend moins de 45 secondes pour les listes de jusqu’à 1 000 gènes (Figure 2, les nombres entre parenthèses représentent les valeurs r2). Ces résultats démontrent la puissance et l’efficacité d’une approche intégrée pour l’annotation fonctionnelle de grands ensembles de données.,

pour la Sortie de l’Outil d’Annotation. Les annotations ajoutées pour les premiers ensembles de sondes Affymetrix dans un tableau HTML contenant les 402 entrées sont affichées. Des informations catégoriques sur les conditions expérimentales ont été soumises avec les identificateurs Affymetrix probe-set et incluses dans la sortie dans la colonne Valeur. Les identifiants tels que les accessions Symbol, LocusLink, OMIM, RefSeq et Unigene sont hyperliés à leurs sources d’origine pour des informations plus détaillées., Le texte inclus dans les champs récapitulatifs est dérivé des informations descriptives et fonctionnelles fournies dans les rapports LocusLink de NCBI.
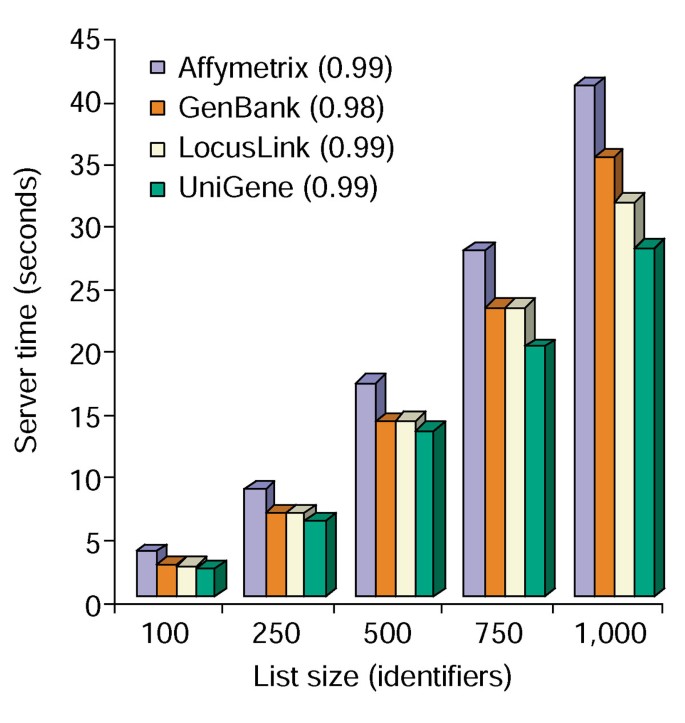
de Temps à l’analyse de l’Outil d’Annotation. Temps de serveur requis (axe y) pour ajouter simultanément les 10 options d’annotation aux listes de gènes dont la taille varie de 100 à 1 000 (axe x)., La moyenne de trois essais pour les listes de gènes contenant des identifiants Affymetrix, GenBank, LocusLink et Unigene est affichée et les nombres entre parenthèses représentent la valeur r2 de la corrélation entre la taille de la liste de gènes et le temps de serveur requis pour l’annotation.
GoCharts
le Choix de la GoCharts module ouvre une nouvelle fenêtre avec une variété d’options., Les utilisateurs choisissent entre trois types généraux de classification (processus biologique, fonction moléculaire et composante cellulaire) et cinq niveaux d’annotation qui représentent la couverture et la spécificité du terme (voir la section Modules D’analyse). Toute combinaison de classification et de niveau de couverture peut être spécifiée. Sont également inclus des options pour annoter des listes de gènes avec tous les Termes GO disponibles ou seulement les termes les plus spécifiques, qui sont appelés nœuds terminaux., La possibilité de choisir différents niveaux de spécificité à terme offre la flexibilité nécessaire et permet ainsi aux chercheurs de déterminer dynamiquement quel niveau de couverture et de spécificité convient le mieux à leurs données et à leur stade d’analyse. Par exemple, les analyses à un stade précoce peuvent consister à annoter des listes de gènes avec des termes très généraux afin d’obtenir une large compréhension des données. Dans ce cas, la sélection du processus biologique et du niveau 1 classe les gènes en utilisant des termes généraux tels que « mort » et « communication cellulaire »., L’utilisation d’une spécificité de terme accrue facilite l’extraction d’informations fonctionnelles plus détaillées. Dans ce cas, la sélection du processus biologique et le niveau 5 classifie les gènes en utilisant des termes tels que « changements mitochondriaux apoptotiques » et « perception chimiosensorielle ».
cependant, l’augmentation de la spécificité des Termes a un coût, en ce sens qu’à mesure qu’elle augmente, la couverture des listes diminue (Figure 3). Dans nos études, nous constatons que le niveau 2 maintient généralement une bonne couverture tout en fournissant une spécificité de terme significative., La Figure 4a illustre comment la visualisation de GoCharts révèle rapidement que 35 gènes exprimés de manière différentielle sont impliqués dans les « réponses au stress ». Chaque terme GO peut être visualisé dans l’arborescence ou les vues DAG par des hyperliens vers QuickGO .
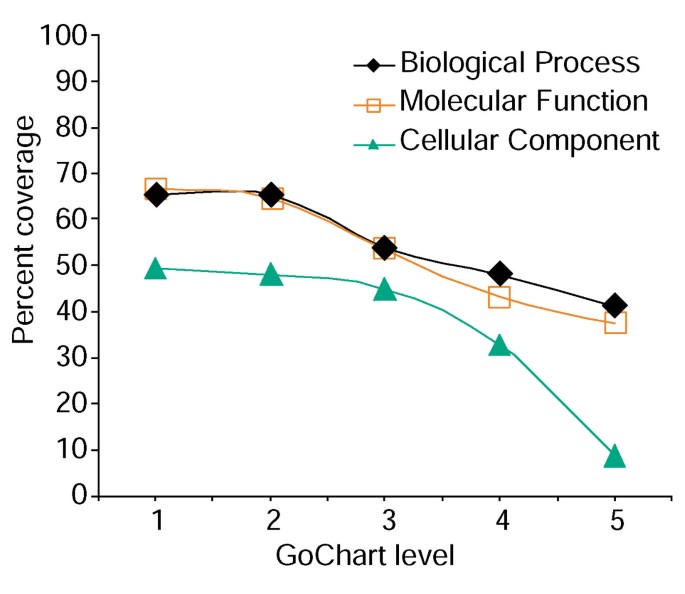
l’Analyse du gène de liste de la couverture à l’aide de GoCharts. Une liste de 402 identificateurs D’ensemble de sondes Affymetrix a été annotée avec les classifications fonctionnelles assignées au protéome fournies par LocusLink., Le pourcentage de couverture représente le nombre de gènes sur 402 qui ont été annotés à un niveau de spécificité de terme dans les types de classification des processus biologiques, des fonctions moléculaires et des composants cellulaires. Le pourcentage de couverture diminue à mesure que la spécificité à terme augmente.
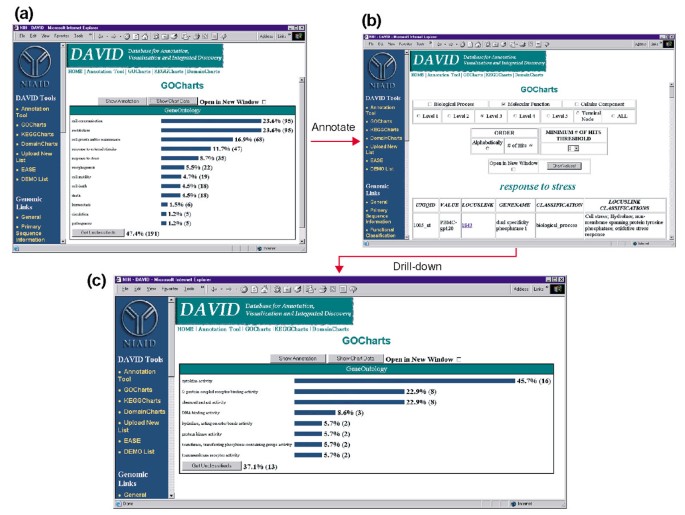
pour la Sortie de GoCharts. (a) un diagramme à barres montrant la distribution des gènes exprimés de façon différentielle entre les processus biologiques de L’ontologie génique (GO)., Les paramètres ont été définis pour aller au niveau 2, un seuil de succès de cinq, et la sortie a été triée par nombre de succès. Les barres bleues sont liées aux données d’annotation supplémentaires indiquées dans (b). La sélection de la barre bleue dans (a) correspondant à « réponse au stress » ouvre un tableau HTML montrant le LocusLink, le nom du gène, la classification actuelle et d’autres données de classification pour les gènes de cette catégorie. (c) ce sous-ensemble de gènes impliqués dans la « réponse au stress » a été caractérisé en sélectionnant la fonction moléculaire GO, le niveau GO 3, un seuil de succès de 2, et trié par nombre de succès., La sélection du bouton » valeurs du graphique » crée un nouvel histogramme révélant que 16 des 35 gènes de réponse au stress codent des protéines possédant une activité cytokine.,
étant donné que le VIH-1 a un impact majeur sur la fonction des cellules du système immunitaire et leur capacité à effectuer des réponses au stress, nous avons sélectionné la barre d’histogramme représentant le nombre de gènes impliqués dans la réponse au stress, qui ouvre un tableau HTML contenant L’identifiant Affymetrix, le numéro de LocusLink, le nom du gène, la classification actuelle et d’autres classifications pour les 35 gènes (Figure 4b)., Maintenant que nous avons réduit notre liste de gènes aux gènes impliqués dans les réponses au stress, nous avons caractérisé ce sous-ensemble en répétant la procédure de GoCharts disponible en haut du tableau HTML stress-réponse. En choisissant la fonction moléculaire, le niveau 3 produit un nouvel histogramme qui révèle rapidement que près de la moitié (16/35) des gènes de réponse au stress possèdent une activité cytokine (Figure 4c)., En effet, il a été démontré que les cytokines jouent un rôle important dans le cycle de vie du VIH-1 et les résultats obtenus ici suggèrent que le traitement des PBMC avec des protéines D’enveloppe du VIH-1 module significativement la transcription de nombreux gènes de cytokines. L’efficacité avec laquelle GoCharts a systématiquement résumé ce grand ensemble de données avec des visualisations graphiques, tout en restant lié aux données primaires et aux ressources externes a considérablement amélioré le processus de découverte.,
KeggCharts
La Figure 5a représente la sortie des KeggCharts avec un histogramme montrant la distribution des gènes différentiellement exprimés entre les voies biochimiques. Le graphique montre qu’une voie de KEGG de l’apoptose comprend cinq gènes induits par le VIH-1 gp120. La sélection du nom de la voie ouvre la carte de voie biochimique de KEGG correspondante et met en évidence en rouge les gènes différentiellement exprimés fonctionnant dans cette voie (Figure 5b). Dans cette vue, les gènes sont en outre liés à des annotations supplémentaires disponibles via le système de récupération DBGET de KEGG ., Notez que seuls quatre gènes de la voie de L’apoptose de KEGG sont surlignés en rouge, tandis que l’outil KeggCharts a cartographié cinq ensembles de sondes Affymetrix sur la voie de l’apoptose. Cette différence est due au fait que deux des ensembles de sondes Affymetrix ciblent le même gène « TNF-alpha ».
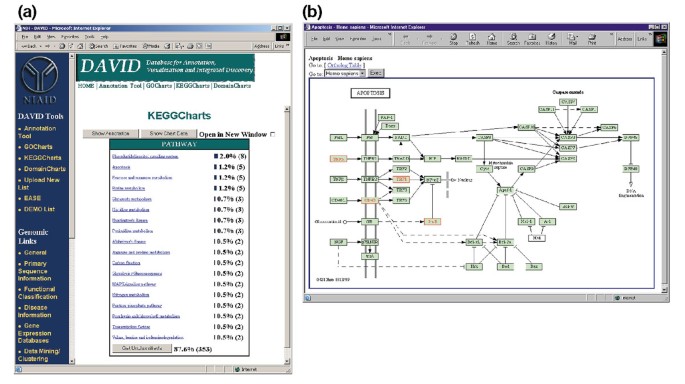
pour la Sortie de KeggCharts. (a) tableau de visualisation montrant la distribution de 402 gènes parmi les voies biochimiques de KEGG. Le seuil d’accès a été défini sur trois et la sortie a été triée par nombre d’accès., Le grand nombre d’identificateurs non classifiés est dû au fait que KEGG est centré sur la voie biochimique et fournit donc une faible couverture des listes de gènes. De même que la sortie des GoCharts, les barres bleues représentent le nombre de gènes dans chaque voie. La sélection d’une barre bleue ouvre un tableau HTML montrant le LocusLink, le nom du gène, la classification actuelle et d’autres données de classification pour les gènes de cette voie (données non affichées)., (b) la voie biochimique de KEGG qui apparaît après la sélection du nom de voie « apoptose » dans (a) représente quatre gènes exprimés différentiellement dans la voie de l’apoptose en les mettant en évidence en vert clair et en rouge. Le fait que la voie de KEGG ne met en évidence que quatre gènes alors que le KeggChart cartographie cinq ensembles de sondes Affymetrix à la voie d’apoptose est dû au fait que deux ensembles de sondes ciblent le même gène « TNF-alpha ».,
diagrammes de domaines
Les diagrammes de domaines s’apparentent fonctionnellement aux diagrammes de Kegg et aux diagrammes de GoCharts, sauf que les résultats décrivent visuellement la distribution des gènes entre les domaines protéiques PFAM (Figure 6a). L’histogramme de DomainCharts identifie 16 gènes avec des domaines kinase (pkinase), reflétant probablement les effets du VIH-1 gp120 sur la machinerie de transduction du signal. Le graphique identifie également six gènes avec des domaines d’interleukine – 8 (IL-8), un domaine qui représente un motif hautement conservé parmi les cytokines de réponse au stress., La sélection du nom de domaine « IL8 » ouvre la page CDD (Conserved Domain Database) correspondant à ce domaine PFAM (Figure 6b). Cette page fournit des informations détaillées sur la séquence, la structure et la fonctionnalité du domaine IL-8 et des protéines qui le contiennent.
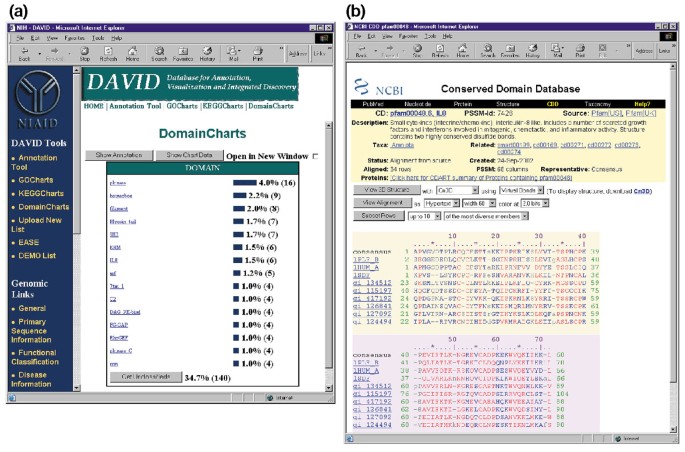
pour la Sortie de DomainCharts. (a) Graphique de visualisation montrant la distribution de 402 gènes entre les domaines protéiques. Les paramètres ont été définis sur un seuil de succès minimum de quatre et la sortie a été triée par nombre de succès., Semblable à la sortie de GoCharts et KeggCharts, les barres bleues représentent le nombre de gènes contenant ce domaine particulier. La sélection d’une barre bleue ouvre un tableau HTML montrant le LocusLink, le nom du gène, la classification actuelle et d’autres données de classification pour les gènes de cette voie (données non affichées)., (b) La sélection du nom de domaine « IL8 » dans (a), qui contient six gènes exprimés de manière différentielle, amène l’utilisateur à une nouvelle page contenant la sortie de la base de données de domaine conservée (CDD) de NCBI, qui fournit des informations détaillées sur le domaine IL-8, y compris des informations structurelles, des alignements de séquences multiples et des informations descriptives