un procedimiento automatizado escrito en Microsoft Visual Basic (VB) 6.,0 actualiza DAVID weekly con los siguientes procedimientos: llamar a una serie de aplicaciones Perl y Java que descargan datos públicos a través de protocolos de transferencia de archivos anónimos (FTP) (Tabla 1); desempaquetar y analizar los datos de anotación deseados; crear archivos de datos delimitados por tabuladores listos para importar la base de datos; e importar datos a un sistema de administración de bases de datos relacionales Oracle 8i (RDBMS) utilizando la aplicación SQL*Loader de Oracle. El servidor web IIE de Microsoft y la tecnología de Página del servidor activo se utilizan para acceder a la base de datos utilizando JavaBeans y el lenguaje de consulta estructurado (SQL)., Los números de LocusLink para los conjuntos de sondas Affymetrix se derivan de Asociaciones de la Universidad de Michigan o NetAffx . Las anotaciones funcionales y las referencias cruzadas de la base de datos se derivan de LocusLink, que proporciona representaciones estables y curadas por humanos de los genes. Para obtener información más detallada sobre las fuentes de datos utilizadas por DAVID, consulte la sección de preguntas frecuentes en .,
módulos de análisis
DAVID se compone de cuatro módulos principales: Herramienta de anotación, GoCharts, KeggCharts y DomainCharts. La herramienta de anotación es un método automatizado para la anotación funcional de listas de genes. Cualquier combinación de datos de anotación se puede elegir entre 10 opciones seleccionando las casillas de verificación apropiadas (Tabla 2)., Las anotaciones se añaden a la lista de genes enviados seleccionando el botón upload, que devuelve una tabla HTML que contiene la lista original del usuario de identificadores anexados con las anotaciones funcionales elegidas. Los genes no anotados se incluyen en la salida sin datos adjuntos para fines de seguimiento.,
El módulo GoCharts muestra gráficamente la distribución de genes expresados diferencialmente entre categorías funcionales utilizando el vocabulario controlado del Gene Ontology Consortium (GO), que proporciona un lenguaje estructurado que se puede aplicar funciones de los genes y proteínas en todos los organismos, incluso cuando el conocimiento continúa acumulándose y cambiando ., El lenguaje está estructurado en un grafo acíclico dirigido (DAG), en el que la especificidad del término aumenta y la cobertura del genoma disminuye a medida que uno se mueve hacia abajo en la jerarquía. En contraste con una verdadera jerarquía, los Términos hijos en un DAG pueden tener más de un término padre y pueden tener una clase diferente de relación con sus diferentes padres. La estructura de GO comienza con tres categorías principales, proceso biológico, función Molecular y componente celular., El proceso biológico incluye objetivos biológicos amplios, como la mitosis o el metabolismo de las purinas, que se logran mediante conjuntos ordenados de funciones moleculares. La función Molecular describe las tareas realizadas por los productos génicos individuales; ejemplos son el factor de transcripción y la helicasa de ADN. El tipo de clasificación de componentes celulares involucra estructuras subcelulares, ubicaciones y complejos macromoleculares; ejemplos incluyen núcleo, telómero y complejo de reconocimiento de origen., Después de elegir un tipo de clasificación, los niveles que determinan la cobertura y la especificidad de la lista se eligen seleccionando el botón de opción apropiado. El nivel 1 proporciona la cobertura más alta de la lista con la menor cantidad de especificidad de término. Con cada nivel creciente, la cobertura disminuye mientras que la especificidad aumenta, de modo que el nivel 5 proporciona la menor cantidad de cobertura con la especificidad de término más alta.
Los datos de clasificación se muestran como un gráfico de barras, donde la longitud de la barra representa el número de identificadores de genes en cada categoría., El usuario puede establecer parámetros de visualización para ordenar los datos de salida y mostrar categorías que contengan al menos un número mínimo de genes. Al seleccionar una barra individual se abre una nueva tabla HTML que muestra el identificador del gen, el número de LocusLink, el nombre del gen, la clasificación actual y otras clasificaciones para cada gen en esa categoría. Un botón » Mostrar todo «abre una nueva tabla HTML que muestra todos los datos de clasificación y un botón» Mostrar datos del gráfico » abre una tabla HTML que contiene los datos del gráfico subyacentes, lo que permite a los usuarios recrear gráficos de gráficos personalizados en un programa de hoja de cálculo., Se puede mostrar un nuevo gráfico para cualquier subconjunto de genes seleccionando el tipo de clasificación y el nivel utilizando las casillas de verificación y los botones de opción disponibles en la página actual del usuario que permiten capacidades de desglose. Un recuento del número de genes anotados se incluye en la salida, y los genes no anotados se agrupan en la categoría «no clasificada», proporcionando así a los usuarios un sistema de seguimiento automatizado para los genes no anotados.
Los KeggCharts muestran gráficamente la distribución de genes expresados diferencialmente entre las vías bioquímicas de KEGG., Cada vía está vinculada al mapa de vías de KEGG, donde los genes expresados diferencialmente de la lista original se resaltan en rojo. En esta vista, los genes están más vinculados a anotaciones adicionales disponibles a través del sistema de recuperación DBGET de KEGG . Al igual que con GoCharts, el usuario puede establecer parámetros de visualización para ordenar los datos de salida y mostrar categorías que contengan al menos un número mínimo de genes y la visualización de KeggCharts hereda todas las características dinámicas de GoCharts.
Los gráficos de dominio muestran la distribución de genes expresados diferencialmente entre los dominios de la proteína PFAM ., Cada designación de dominio está vinculada a la base de datos de dominios conservados (CDD) del Centro Nacional de Información Biotecnológica (NCBI), donde los detalles relativos a la función, estructura y secuencia del dominio están fácilmente disponibles. Al igual que con GoCharts y KeggCharts, el usuario puede establecer parámetros de visualización para ordenar los datos de salida y mostrar categorías que contienen al menos un número mínimo de genes y la visualización de DomainCharts hereda todas las características dinámicas de GoCharts y KeggCharts. Para obtener más información sobre la funcionalidad de DAVID, visite la sección de preguntas frecuentes en .,
usando DAVID para minar la anotación funcional
para demostrar la funcionalidad de DAVID analizamos una lista de genes expresados diferencialmente en células mononucleares de sangre periférica humana (PBMCs) después de la incubación con proteínas de la envoltura del VIH-1. Los detalles de los procedimientos experimentales, de preparación de ARN y de hibridación GeneChip, junto con los detalles de las normalizaciones chip-a-chip y el análisis estadístico de la expresión génica diferencial se proporcionan en Cicala et al. ., Brevemente, las PBMCs humanas primarias y los macrófagos derivados de monocitos se incubaron durante 16 horas con la proteína de la envoltura del VIH-1 (gp120). Se utilizaron microarrays de oligonucleótidos de alta densidad (Affymetrix HU-95a GeneChip) para monitorear eventos transcripcionales inducidos por gp120. Este análisis resultó en la identificación de 402 genes expresados diferencialmente.
mientras que 16 genes modulados por el VIH-1 gp120 se han asociado previamente con la replicación del VIH y/o la señalización de la envoltura, los genes restantes son de función desconocida o nunca se han asociado con el VIH-1 o gp120., Convertir esta lista de genes en significado biológico requiere la recopilación de información pertinente de varios repositorios de datos. Para muchos investigadores, este proceso consiste en navegar iterativamente a través de varias bases de datos para cada gen, recopilando manualmente información específica del gen con respecto a la secuencia, la función, la Vía y la Asociación de la enfermedad. En contraste, el enfoque sistemático de DAVID agrega simultáneamente información biológicamente Rica derivada de varias fuentes de datos públicos a listas de genes en paralelo., Seleccionar la herramienta de anotación de DAVID y cargar la lista de 402 genes expresados diferencialmente inicia la anotación funcional y el análisis de todo el conjunto de datos. Una vez enviada, la lista de genes se almacena durante toda la sesión de análisis, lo que permite a los usuarios cambiar entre módulos sin tener que volver a enviar datos.
herramienta de anotación
la herramienta de anotación proporciona varias opciones de anotación y construye una vista tabular de la lista de genes de los usuarios y las anotaciones disponibles (Tabla 2)., La elección de los campos de anotación Gene Symbol, LocusLink, OMIM, UNIGENE, Reference Sequence y Gene Name seguido de la selección del botón «Upload» produce una tabla HTML en el navegador web que contiene todos los genes y sus anotaciones disponibles, donde los identificadores de genes, los datos descriptivos y de clasificación se extraen de la base de datos y se anexan a la lista de genes (Figura 1). Los identificadores de genes como el símbolo del gen y el LocusLink están hipervinculados a datos específicos de genes adicionales disponibles en sus fuentes originales, proporcionando así detalles específicos de genes en profundidad y pedigríes de anotación., Los datos de clasificación y los resúmenes funcionales se pueden utilizar para escanear rápidamente información relevante para el sistema experimental del investigador. El tiempo del servidor requerido para la ejecución de este módulo se correlaciona linealmente con el tamaño de la lista de genes y toma menos de 45 segundos para las listas de hasta 1.000 genes (Figura 2, los números entre paréntesis representan los valores r2). Estos resultados demuestran el poder y la eficiencia de un enfoque integrado para la anotación funcional de grandes conjuntos de datos.,

la Salida de la Herramienta de Anotación. Se muestran anotaciones anexas para los primeros conjuntos de pruebas de Affymetrix en una tabla HTML que contiene las 402 entradas. La información categórica sobre las condiciones experimentales se presentó junto con los identificadores Affymetrix probe-set y se incluyó en la salida en la columna Valor. Los identificadores como las accesiones Symbol, LocusLink, OMIM, RefSeq y Unigene están hiperenlazados a sus fuentes de origen para obtener información más detallada., El texto incluido en los campos de resumen se deriva de la información descriptiva y funcional proporcionada en los informes LocusLink del NCBI.
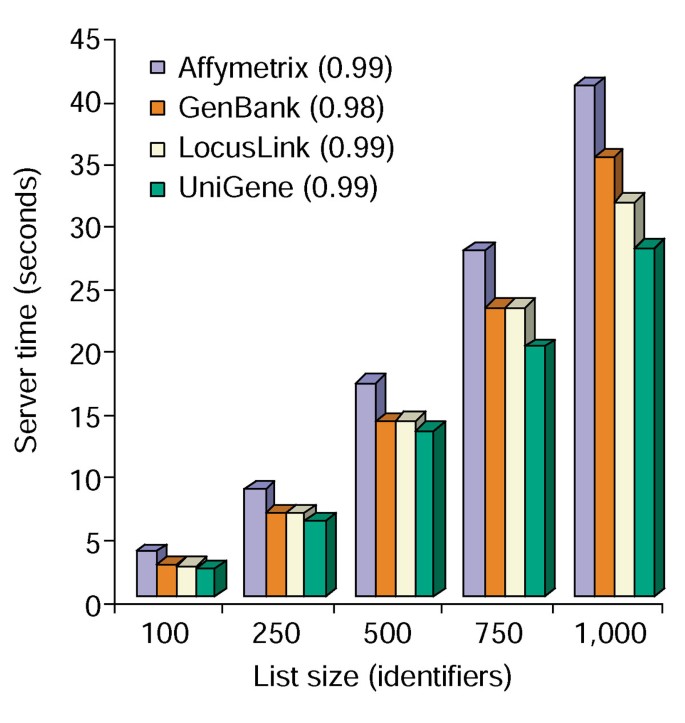
Tiempo de análisis de la Herramienta de Anotación. Se requiere tiempo del servidor (eje y) para anexar simultáneamente las 10 opciones de anotación a listas de genes que varían en tamaño de 100 a 1.000 (eje x)., Se muestra el promedio de tres ensayos para listas de genes que contienen identificadores Affymetrix, GenBank, LocusLink y UniGene y los números entre paréntesis representan el valor r2 de la correlación entre el tamaño de la lista de genes y el tiempo del servidor requerido para la anotación.
GoCharts
al elegir el módulo GoCharts se abre una nueva ventana con varias opciones., Los usuarios eligen entre tres tipos generales de clasificación (proceso biológico, función molecular y componente celular) y cinco niveles de anotación que representan la cobertura y especificidad del término (ver sección módulos de análisis). Se puede especificar cualquier combinación de clasificación y nivel de cobertura. También se incluyen opciones para anotar listas de genes con todos los Términos GO disponibles o solo los términos más específicos, que se conocen como nodos terminales., La opción de elegir diferentes niveles de especificidad de término proporciona la flexibilidad necesaria y, por lo tanto, permite a los investigadores determinar dinámicamente qué nivel de cobertura y especificidad se adapta mejor a sus datos y etapa de análisis. Por ejemplo, los análisis en las primeras etapas pueden consistir en anotar listas de genes con Términos muy generales con el fin de obtener una amplia comprensión de los datos. En este caso, la selección de proceso biológico y Nivel 1 clasifica los genes Utilizando términos generales como » muerte «y»comunicación celular»., El uso de una mayor especificidad de término facilita la extracción de información funcional más detallada. En este caso la selección de proceso biológico y nivel 5 clasifica los genes Utilizando términos como «cambios mitocondriales apoptóticos» y «percepción quimiosensorial».
sin embargo, el aumento de la especificidad del término tiene un costo, ya que a medida que aumenta la cobertura de la lista disminuye (Figura 3). En nuestros estudios encontramos que el nivel 2 típicamente mantiene una buena cobertura al mismo tiempo que proporciona una especificidad de término significativa., La figura 4a ilustra cómo la visualización de GoCharts revela rápidamente que 35 genes expresados diferencialmente están involucrados en las»respuestas al estrés». Cada término GO se puede ver en las vistas de árbol o DAG mediante hipervínculos a QuickGO .
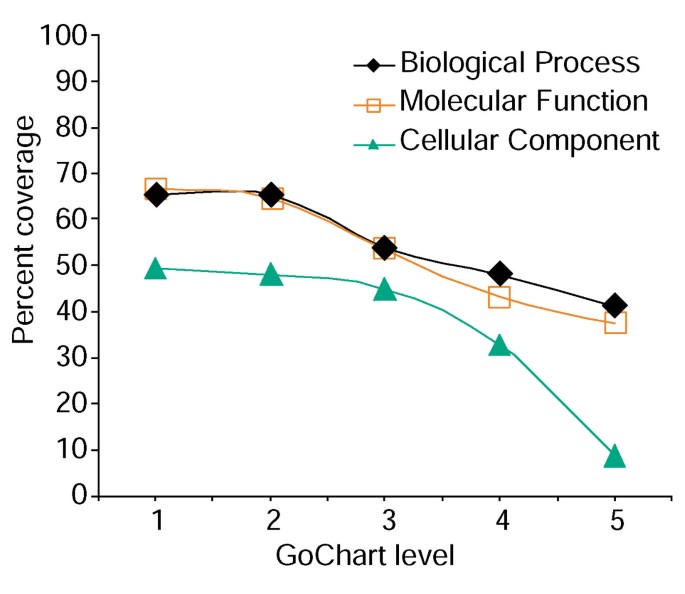
el Análisis del gen de la lista de cobertura utilizando GoCharts. Se anotó una lista de 402 identificadores del conjunto de sondas Affymetrix con las clasificaciones funcionales asignadas al proteoma proporcionadas por LocusLink., La cobertura porcentual representa el número de genes de 402 que fueron anotados a un nivel de especificidad de término dentro de los tipos de clasificación de procesos biológicos, funciones moleculares y componentes celulares. La cobertura porcentual disminuye a medida que aumenta la especificidad del término.
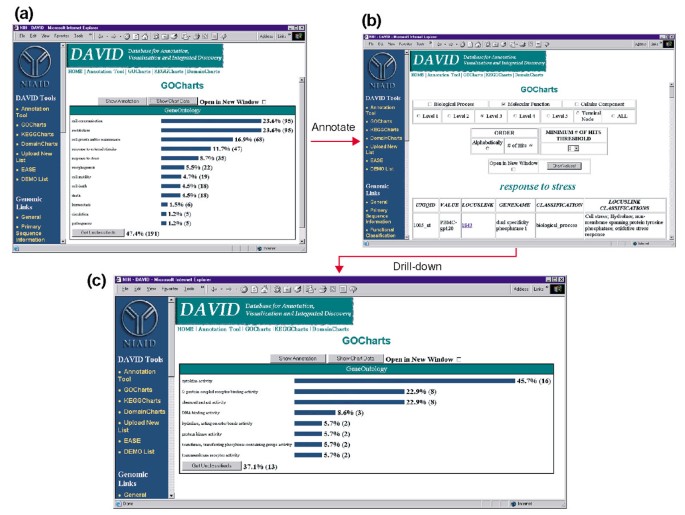
la Salida de GoCharts. (a) un gráfico de barras que muestra la distribución de genes expresados diferencialmente entre los procesos biológicos de Ontología génica (GO)., Los parámetros se establecieron en el nivel 2, un umbral de acierto de cinco, y la salida se clasificó por número de aciertos. Las barras azules están vinculadas a datos de anotación adicionales que se muestran en (b). Al seleccionar la barra azul en (a) correspondiente a «respuesta al estrés» se abre una tabla HTML que muestra el LocusLink, el nombre del gen, la clasificación actual y otros datos de clasificación para los genes en esa categoría. (c) este subconjunto de genes involucrados en la «respuesta al estrés» se caracterizó además seleccionando la función Molecular GO, el nivel GO 3, un umbral de hit de 2, y se clasificó por recuento de hit., Al seleccionar el botón «valores del gráfico» se crea un nuevo histograma que revela que 16 de los 35 genes de respuesta al estrés codifican proteínas que poseen actividad de citoquinas.,
debido a que el VIH-1 tiene un impacto importante en la función de las células del sistema inmunológico y su capacidad para llevar a cabo respuestas al estrés, se seleccionó la barra de histograma que representa el número de genes involucrados en la respuesta al estrés, que abre una tabla HTML que contiene el identificador Affymetrix, el número de LocusLink, el nombre del gen, la clasificación actual y otras clasificaciones para los 35 genes (figura 4b)., Ahora que hemos reducido nuestra lista de genes a aquellos genes involucrados en las respuestas al estrés, caracterizamos este subconjunto repitiendo el procedimiento de GoCharts disponible en la parte superior de la tabla HTML de respuesta al estrés. Al elegir la función molecular, el Nivel 3 produce un nuevo histograma que revela rápidamente que casi la mitad (16/35) de los genes de respuesta al estrés poseen actividad de citoquinas (figura 4c)., De hecho, se ha demostrado que las citoquinas desempeñan un papel importante en el ciclo de vida del VIH-1 y los resultados obtenidos aquí sugieren que el tratamiento de PBMCs con proteínas de la envoltura del VIH-1 modula significativamente la transcripción de numerosos genes de citoquinas. La eficiencia con la que gocharts resumió sistemáticamente este gran conjunto de datos con visualizaciones gráficas, mientras permanecía vinculado a datos primarios y recursos externos mejoró drásticamente el proceso de descubrimiento.,
KeggCharts
La Figura 5a representa la salida de KeggCharts con un histograma que muestra la distribución de genes expresados diferencialmente entre las vías bioquímicas. El gráfico muestra que una vía KEGG de apoptosis incluye cinco genes inducidos por el VIH-1 gp120. Al seleccionar el nombre de la Vía se abre el correspondiente Mapa de la vía bioquímica de KEGG y se resaltan en rojo los genes expresados diferencialmente que funcionan en esa vía (figura 5b). En esta vista, los genes están más vinculados a anotaciones adicionales disponibles a través del sistema de recuperación DBGET de KEGG ., Tenga en cuenta que solo cuatro genes en la vía de apoptosis de KEGG están resaltados en rojo, mientras que la herramienta KeggCharts mapeó cinco conjuntos de sonda Affymetrix a la vía de apoptosis. Esta diferencia se debe al hecho de que dos de los probesets Affymetrix se dirigen al mismo gen «TNF-alfa».
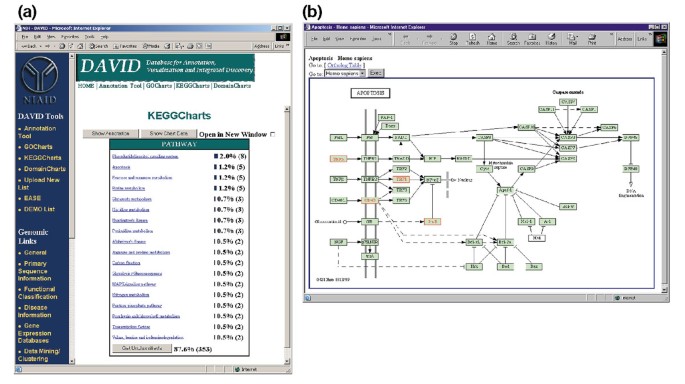
la Salida de KeggCharts. a) gráfico de visualización que muestra la distribución de 402 genes entre las vías bioquímicas de KEGG. El umbral de visitas se estableció en tres y la salida se clasificó por número de visitas., El gran número de identificadores no clasificados se debe al hecho de que KEGG está centrado en la vía bioquímica y, por lo tanto, proporciona una baja cobertura de las listas de genes. De manera similar a la salida de GoCharts, las barras azules representan el número de genes en cada vía. Al seleccionar una barra azul se abre una tabla HTML que muestra el LocusLink, el nombre del gen, la clasificación actual y otros datos de clasificación para los genes en esa ruta (datos no mostrados)., (b) La vía bioquímica de KEGG que aparece después de la selección del nombre de la vía «apoptosis» en (A) representa cuatro genes expresados diferencialmente dentro de la vía de apoptosis resaltándolos en verde claro y rojo. El hecho de que la vía de KEGG resalta solo cuatro genes, mientras que el KeggChart mapea cinco conjuntos de sonda Affymetrix a la vía de apoptosis se debe al hecho de que dos conjuntos de sonda apuntan al mismo gen «TNF-alfa».,
DomainCharts
DomainCharts son operacionalmente similares a los KeggCharts y GoCharts, excepto que los resultados muestran visualmente la distribución de genes entre los dominios de la proteína PFAM (figura 6a). El histograma DomainCharts identifica 16 genes con dominios de quinasa (pkinasa), probablemente reflejando los efectos del VIH-1 gp120 en la maquinaria de transducción de señales. El gráfico también identifica seis genes con dominios de interleucina-8 (IL-8), un dominio que representa un motivo altamente conservado entre las citocinas de respuesta al estrés., Al seleccionar el nombre de dominio «IL8» se abre la página de base de datos de dominios conservados (CDD) correspondiente a ese dominio PFAM (figura 6b). Esta página proporciona información detallada sobre la secuencia, la estructura y el funcionamiento del dominio IL-8 y las proteínas que lo contienen.
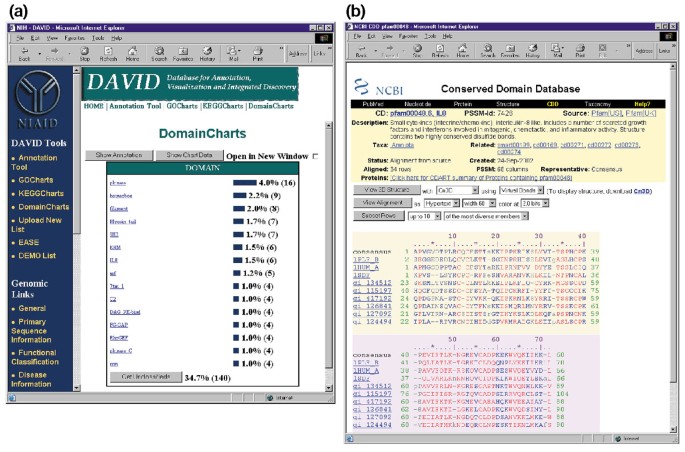
la Salida de DomainCharts. (a) gráfico de visualización que muestra la distribución de 402 genes entre dominios proteicos. Los parámetros se establecieron en un umbral mínimo de aciertos de cuatro y la salida se clasificó por recuento de aciertos., Similar a la salida de GoCharts y KeggCharts, las barras azules representan el número de genes que contienen ese dominio particular. Al seleccionar una barra azul se abre una tabla HTML que muestra el LocusLink, el nombre del gen, la clasificación actual y otros datos de clasificación para los genes en esa ruta (datos no mostrados)., (b) La Selección del nombre de dominio «IL8» en (a), que contiene seis genes expresados diferencialmente, lleva al usuario a una nueva página que contiene la salida de la base de datos de dominios conservados (CDD) de NCBI, que proporciona información detallada sobre el dominio IL-8, incluida información estructural, alineaciones de secuencias múltiples e información descriptiva sobre el dominio y las proteínas que lo poseen.