el gen p53 se descubrió por primera vez en 1979. Se identificó una proteína en células de ratón del virus simio 40 transformadas (SV40) por inmunoprecipitación con Suero anti-T; esta proteína se llamó proteína p53 . En el mismo año, Kress y otros científicos encontraron una nueva clase de proteínas con una masa molecular que va desde 50-60kDa. Este tipo de proteína fue identificado como p53 . La proteína p53 también se puede identificar a partir de varias líneas celulares transformadas por inmunoprecipitación., Lane y Linzer también obtuvieron un resultado similar en 1979. Otra evidencia para identificar p53 es que p53 se expresó en todas las células transformadas de ratón probadas; estas pruebas incluyen sarcomas inducidos químicamente, fibroblastos transformados y leucemias, mientras que en las células normales, p53 no se expresó. Además, se detectó un alto nivel de p53 en la mayoría de las células transformadas, sin importar cómo se transformaron las células, ya sea espontáneamente o no espontáneamente ., Eso fue en gran parte debido a la mayor estabilidad de la p53, sin embargo, en células embrionarias de carcinoa F9, expresó un alto nivel de p53, esto se debió a la cantidad de mRNA p53 traducido .
después de que la proteína p53 hubiera sido descubierta en 1979, se hizo popular analizarla. Sin embargo, en ese momento, como era una proteína recién descubierta, y no había un nombre anterior para ella, diferentes instituciones usaron diferentes nombres y publicaron artículos con diferentes nombres., Con el fin de resolver este problema, en 1983, durante el 1st International p53 workshop celebrado en Oxted, Reino Unido, científicos de diferentes grupos de investigación en diferentes países se reunieron para discutir una nomenclatura común para esta proteína recién descubierta. En esta reunión,» p53 » se convirtió en su nombre y se ha utilizado desde entonces. Se pensó que la razón por la que los científicos llamaron a la proteína p53 es que la masa molecular de esta proteína es de 53kDa que se basa en su migración en gel SDS. Más tarde se demostró que la masa molecular era incorrecta, y la masa molecular correcta debería ser 43.,7 kd porque p53 contiene una región rica en prolina, y esta región puede reducir la migración de p53 en gel de SDS. Pero el nombre «p53» se mantuvo .
durante la década de 1980, se creía que la proteína p53 estaba involucrada en el ciclo celular, además de desempeñar un papel en la replicación del ADN. Más tarde, en 1982 a 1994, la gente encontró que algunas oncoproteínas virales eran capaces de unirse a p53, formando un complejo. En 1982, Sarnow et al. se encontró que el adenovirus E1B (58kDa) puede interactuar con una proteína 54kDa que está presente en las células de ratón transformadas por SV40 mencionadas anteriormente., De acuerdo con los resultados de las especificidades inmunológicas de los anticuerpos T y los mapas peptídicos de la proteína 54kDa, esta proteína 54kDa se identifica como p53 . En el mismo año, los científicos encontraron que si inyectaban el anticuerpo p53 en células de ratón Swiss 3T3 inhibiría las células que entraban en la fase S del ciclo celular, sin embargo; bajo la misma situación, el anticuerpo p53 no afectó SV40 o la síntesis de ADN inducida por adenovirus .,
Más tarde, en 1984, los científicos examinaron el efecto del p53 en los fibroblastos 3T3 no transformados; analizaron la tasa de síntesis de la proteína p53 en diferentes puntos de tiempo y encontraron que en la fase G1 tardía, la tasa de síntesis y el nivel de la proteína p53 y su aumento relacionado del ARNm. Este resultado sugiere que la proteína p53 inhibe las células que entran en la fase de división de la interfase . Maltzman W y otros hizo otro experimento en el mismo año. Trataron la célula de ratón no transformada con luz UV y carcinógeno químico UV-mimético 4NQO, y detectaron un alto nivel de p53., El resultado mostró que la expresión elevada de p53 no es solo un símbolo que indica el ciclo celular, sino también, lo que es más importante, un componente que está involucrado en la síntesis de ADN y la proliferación celular . En 1987, al estudiar el complejo del antígeno T del virus simio 40 y la ADN polimerasa α, Gannon y otros científicos encontraron un cambio similar en el antígeno cuando se une a p53 y polimerasa α. También encontraron que a una cierta concentración de los tres componentes, pueden formar un complejo trimérico especial que incluye antígeno T, P53 y ADN polimerasa α., Como el antígeno T está involucrado en la replicación del ADN viral y la transformación celular, este resultado indica que p53 juega un papel en el control del ciclo celular y la replicación del ADN .
como el experimento mostró anteriormente, p53 tiene la capacidad de inmortalizar células. En 1984, Eliyahu D et al. encontró que p53 y el producto de oncogene myc compartían algunas propiedades similares. Ambos tienen la capacidad de unirse a otras proteínas y están involucrados en el ciclo celular, y ambos se acumulan en núcleos de células transformadas ., Bienz, Pennica y Oren analizaron las secuencias de aminoácidos de la proteína p53 y el producto de myc, y encontraron que las dos proteínas muestran similitudes en la estructura molecular y la posición de los residuos cargados especiales. Entonces los científicos propusieron que el p53 podría actuar como un oncogén. Con base en esta hipótesis, Eliyahu D et al. hice algunos experimentos. Como los fibroblastos embrionarios de tasa primaria pueden ser transformados por la participación del producto myc y ha-ras, las células primarias de riñón de rata bebé también pueden ser transformadas por la cooperación de Ha-ras y adenovirus región temprana 1A , Eliyahu D et al., decidió utilizar este tipo de sistema de pruebas biológicas para identificar la función oncogénica del p53. Trataron células embrionarias normales con p53 y activaron Ha-ras. El resultado mostró que las células diana encuentran cambios morfológicos y producen altos niveles de p53, Eliyahu D et al. pensó que la transformación de los fibroblastos embrionarios por p53 y ha-ras explicó que el gen p53 es un oncogén ., En 1985, Jenkins propuso que el gen p53 puede extender la vida útil de las células, mejorar la afectividad de la transformación mediante la reorganización de su secuencia de codificación que podría causar la producción de proteínas estables .
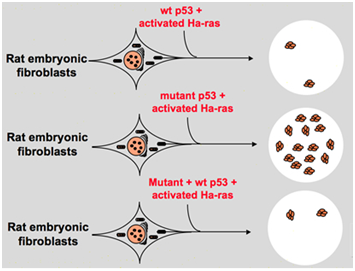
sin embargo, a finales de la década de 1980, los científicos comenzaron a darse cuenta de que p53 es un gen supresor de tumores en lugar de un oncogén. Observaron que p53 con función normal no se puede detectar en muchos de los tumores y encontraron que la pérdida de la expresión y la función del gen p53 de tipo salvaje es necesaria durante la transformación celular., Estos aumentan la posibilidad de que el gen p53 de tipo salvaje pueda inhibir la progresión neoplásica . Luego formularon otra hipótesis: el gen clon p53 utilizado en experimentos anteriores contiene mutaciones negativas dominantes dentro del dominio altamente conservado ocasionalmente, que conducen a resultados de experimentos opuestos . En 1988, Ben y otros científicos detectaron una gran cantidad de p53 reorganizado en líneas celulares de eritroleucemia murina DP DP20-1 y CB3 que se derivan de los bazos de Murino infectados con el virus de la leucemia Friend ., En 1989, Eliyahu, quien señaló que p53 es un oncogén, cambió de opinión, y supuso que el gen p53 de tipo salvaje puede inhibir la transformación celular. Eliyahu y otros científicos estudiaron el efecto de la proteína p53 de tipo silvestre codificada por plásmidos y p53 mutante en la capacidad de provocar la transformación de fibroblastos embrionarios de tasa primaria por varias combinaciones de oncogenes in vitro. Por ejemplo mutante p53 más ras, y myc más ras., El resultado mostró que el tipo salvaje p53 conduce a una enorme reducción de los focos transformados causados por el mutante p53 más ras; El Mutante p53 no mostró inhibición en los focos transformados causados por myc más ras, mientras que la transformación mediada por myc más ras es muy sensible a la expresión del tipo salvaje p53. La figura 1 muestra este experimento de manera concisa. Mostró que en comparación con el mutante p53, el tipo salvaje p53 exhibe un efecto obviamente inhibitorio en la transformación celular. El efecto se relaciona positivamente con el nivel de expresión de p53 Salvaje y negativamente con el nivel de expresión de p53 mutante., Este experimento sugirió que el p53 de tipo salvaje puede tener una función opuesta en comparación con el p53 mutante y puede inhibir la tumorogénesis . Actualmente, p53 es reconocido como un gen supresor de tumores. Se estima que aproximadamente la mitad de los tumores son causados por p53. Es uno de los genes mutados con más frecuencia en los seres humanos, y el gen analizado con más frecuencia en todo el mundo .
durante los primeros años de la década de 1980, la vía bioquímica de p53 y el efecto de la mutación p53 no estaban claros. En 1991, Kern y otros científicos encontraron que una secuencia de ADN de 33 pares de bases se une específicamente a p53 de tipo salvaje in vitro. También encontraron que la proteína p53 contiene dos mutaciones que generalmente se encuentran en tumores humanos que no pueden unirse a esta región específica del ADN. Así que supusieron que la función de p53 depende de su capacidad para unir secuencias específicas de ADN, y esta capacidad es alterada por mutaciones encontradas en tumores humanos., También suponen que esta secuencia de ADN de 33 pares de bases puede no ser la única secuencia que tiene la capacidad de unirse específicamente al p53 en humanos; sin embargo, puede ayudar a las personas a comprender mejor la función del p53 . Más tarde, se descubrió que p53 desempeñaba un papel durante el ciclo celular, la reparación del ADN, la diferenciación, el inicio de la apoptosis y la angiogénesis. Rotter V et al. se encontró que p53 regula la diferenciación de las células. Por ejemplo, se detectó un alto nivel de proteína p53 en varios pasos clave durante la diferenciación de células B. P53 elevado también se puede detectar durante la espermatogénesis., Mientras tanto, solo se puede detectar un nivel muy bajo de proteína p53 en algunos órganos de ratones adultos .
en 1990, una herramienta útil fue descubierta ocasionalmente. Es un mutante sensible a la temperatura de p53, llamado p53val135. Puede actuar como un p53 de tipo salvaje real a la temperatura de 32.5 oC, suprimiendo la transformación, y también puede actuar como otro p53 mutado a la temperatura de 37.5 oC o por encima de 48oC, provocando la transformación. Además, para las células transformadas que expresan p53val135, su proliferación se controla a la temperatura permisiva, y este tipo de control es reversible., Mediante el uso de este mutante p53val135, se descubrió que p53 de tipo salvaje induce la detención del ciclo celular en G1 o G2 / M . En 1991, Elisheva et al. se encontró que la p53val135 sensible a la temperatura desempeñaba una función diferente en la línea celular de leucemia mieloide murina. Después de la reactivación de p53val135 durante unos días, todas las células murieron, y esta muerte exhibe algunas propiedades de apoptosis . Un año más tarde, un resultado similar fue obtenido por Shaw. Un tipo salvaje p53 fue transfectado en una línea celular EB derivada de un tumor de colon humano., Las células fueron examinadas bajo microscopios de luz y electrónicos, y se encontró que exhiben algunas propiedades de apoptosis . En 1990, Scheffner et al. y otros científicos encontraron que el E6 que estimula la destrucción de las proteínas reguladoras de la célula huésped está codificado por el virus del papiloma humano oncogénico tipos 16 y 18, y puede formar un complejo con p53 de tipo silvestre in vitro, que a su vez causa la degradación de la proteína p53 .
en 1992, se descubrió una proteína clave MDM2 porque se une estrechamente con p53, e inhibe la transactivación mediada por p53., La masa molecular de MDM2 es 90kDa, y forma un complejo con p53 mutado y salvaje . En el mismo año, Livingstone RL et al. se estudió si la célula perdió una o ambas copias de alelos p53 de tipo salvaje y si eso fue suficiente para causar amplificación génica. La amplificación génica se detectó principalmente en células transformadas, pero no en los fibroblastos normales. El resultado mostró que las células que pierden una copia de los alelos p53 actúan como p53 de tipo salvaje, mientras que las células que pierden ambas copias de los alelos p53 de tipo salvaje exhiben una mayor frecuencia de amplificación . Otro experimento realizado por Yin y et al., mostró un resultado similar .
en 1993, se identificó un gen diana p53 llamado CDKN1A. Codifica la proteína p21 que es un inhibidor de la cinasa dependiente de ciclina que inhibe la ciclina-CDK2 y CDK1 al unirse a ellos. En 1993, Szekely descubrió que el antígeno nuclear 5 del virus de Epstein-Barr(EBNA-5) está codificado por el virus de Epstein-Barr, y puede infectar las células linfoblastoides B humanas. Un péptido largo de 66 aminoácidos es responsable de la formación del complejo EBNA-5-p53, las mutaciones puntuales de p53 no afectaron su capacidad de unión a EBNA-5., Sin embargo, inhibe sus formaciones de complejos con otras moléculas . En 1994, Cho y sus compañeros de trabajo describieron por primera vez la estructura cristalina del complejo p53-DNA. Este dominio de unión de ADN también se llamó dominio Central. Contiene residuos 102-292, y consiste en un sándwich beta. También demostraron la estructura detallada del dominio Central . También en 1994, Wang XW et al. la interacción entre la proteína X del virus de la hepatitis B (HBX) y la proteína p53 de tipo silvestre en humanos., Encontraron que el HBX puede inhibir la capacidad de p53 para unirse a otro ADN de secuencia específica después de que se une a p53 y también puede inhibir la Asociación de p53 con factores de transcripción .
en 1997, Honda R et al. first hypothesized that MDM2 can trigger p53 ubiquitylation and lead to degradation of p53 by a ubiquitin-proteasome system. Señalaron que MDM2 se une al dominio N-terminal (NTD) de p53 y actúa como ubiquitina ligasa E3 . También en 1997, dos nuevas familias de proteínas, P63 y p73 fueron descubiertas que comparten homología sustancial con p53., p73, también llamada proteína tumoral 73, es codificada por un gen localizado en 1p36. La ubicación se elimina con frecuencia en el neuroblastoma y otros tumores. p73 puede activar genes diana p53 e interactúa con p53 . Yang et al. se encontró que el gen p63 se encuentra en 3q27 – 29 y se puede detectar en varias células de ratón y humanos. Al igual que p73, p63 puede transactivar los genes diana p53 significativamente, también puede inducir apoptosis. Una característica de p63 es que la mayoría de p63 carece de un N-terminal ., En el mismo año, Serrano y compañeros de trabajo encontraron que los fibroblastos murinos primarios pueden ser transformados por ras en ausencia de p53 o p16, y los p53 o P16 inactivos pueden facilitar el proceso de inmortalización de las células humanas. Estos hallazgos sugieren que p53 juega un papel en la senescencia celular . Luego, en 1997, se encontró que p53 desempeñaba un papel en el inicio de la apoptosis. Cuando las células entran en la fase de proliferación, los telómeros al final de cada cromosoma se acortarían después de cada ronda de replicación del ADN debido a la replicación incompleta del ADN de una sola cadena al final del soporte de ADN ., El gen supresor tumoral activado p53 limita el número de veces que puede ocurrir la división celular. Wynford TD encontró que con la pérdida de la función del tipo salvaje p53, todos los fibroblastos escapan de la apoptosis. Además, la función de transactivación de p53 se puede activar por apoptosis . Wynford TD propuso que hay tres posibilidades de cómo se activa p53. El primero es la modificación post-traslacional por fosforilación, el segundo es arriba-regula los cofactores transcripcionales como p33ING1, el último es abajo-regula los inhibidores p53 como MDM2 .
en 2000, Brodsky MH et al., se estudiaron las dianas de transcripción de p53 en Drosophila. Hay evidencia para mostrar que los ojos de Drosophila muestran un fenotipo severo bajo la expresión de p53 humano que inducirá la apoptosis de las células del disco imaginal del ojo, causando la pérdida de células pigmentarias, finalmente inhibiendo el desarrollo ocular de Drosophila , por lo que Drosophila puede ser un animal modelo para estudiar la función de p53. Brodsky encontró que el gen RPR contiene un sitio de unión de consenso p53 que se encuentra en la región cis-reguladora de rpr, y también es un activador de la apoptosis., Con otras pruebas, Brodsky afirmó que el rpr es un objetivo transcripcional del p53 . En 2001, Derry y compañeros de trabajo encontraron que C. elegans no tiene un gen p53, pero de hecho contienen un gen cep-1 que codifica proteínas que tienen una secuencia similar con la proteína p53. Este gen de C. elegans codifica la proteína CEP-1 que tiene la capacidad de inducir apoptosis por estrés genotóxico y es un componente necesario durante la meiosis .
en 2002, Tyner y compañeros de trabajo propusieron que p53 juega un papel en la regulación del envejecimiento de los organismos., Para estudiar la función de p53, crearon ratones genéticamente modificados con p53 mutado mediante la eliminación de los exones 1-6 y una región aguas arriba del gen p53 de tipo salvaje (p53+/+), llamado p53+/m. actúa como p53 de tipo salvaje y ha aumentado la resistencia a los tumores espontáneos mejor que p53 de tipo salvaje. En el experimento, monitorearon los ratones que contenían p53+ / m, p53+ / + y p53+/ -. p53 + / – significa que los ratones pierden una copia del gen p53 salvaje., Los resultados mostraron que ninguno de los ratones con p53+/m desarrolló tumores potencialmente mortales, sin embargo, más del 80% de los ratones con p53+/- y más del 45% de los ratones con p53+/+ desarrollaron este tipo de tumores. Al observar el interior de los tumores, se observaron lesiones tumorales localizadas en 2 de 35 ratones p53+/m, en contraste, se encontraron varios tumores como linfomas y osteosarcomas en ratones p53+/- y p53+/+. Durante este experimento, también observaron que la mediana de edad de p53+/m fue de 96 semanas, mientras que la mediana de edad de p53+/m fue de 116 a 118 semanas., Tyner y compañeros de trabajo también examinaron la posibilidad de que la vida más corta de p53+/m se asociara con el envejecimiento. Encontraron que después de 18 meses, los ratones p53+/m comenzaron a perder peso y vigor, como para los ratones p53+/m, Se observaron Pesos reducidos a la edad de 30-36 meses. los ratones p53+ / m también exhiben lordocifosis. Dependiendo del análisis de rayos X, los ratones p53+/m mostraron una densidad ósea reducida a la edad de 12 meses, y se volverá grave a la edad de 18 meses. Este es un símbolo de la osteoporosis y la osteoporosis es un marcador del envejecimiento en humanos y ratones . Tyner et al., también se ha probado la tolerancia al estrés, ya que esta capacidad es también un marcador del envejecimiento . Se realizaron biopsias de punzón de 3 mm en la piel posterior de ratones viejos y jóvenes anestesiados p53+/m y p53+/+. Sus resultados mostraron que muchos ratones viejos p53+/m murieron después de inyectar la dosis estándar de Avertin, lo que indica que los ratones viejos p53+/m fueron menos tolerantes al estrés .
en 1991, se encontró que p53 tiene la capacidad de inducir apoptosis, mientras que en 2003; Mihara y otros científicos encontraron que p53 también tiene un papel de apoptosis en las mitocondrias ., Dado que algunas proteínas mitocondriales tienen la capacidad de activar la apoptosis celular ya sea por caspasas activas o neutralizando inhibidores citosólicos. En el ejemplo de la caspasa inducida por el citocromo c, después de recibir la señal de apoptosis, el citocromo c se libera desde el espacio intermembrana de las mitocondrias, y luego a su vez se une al Apf-1 que existe como un monómero inactivo, induce su cambio conformacional y aumenta su afinidad de unión por dATP/ATP en 10 veces que el Apaf-1 se une a dATP/ATP solo. A continuación, el complejo Apaf-1 – citocromo c se une a dATP/ATP, forma el apoptosoma., Después de eso, el dominio de reclutamiento caspasa (CARD) de Apaf-1 expuesto en el apoptosoma, recluta la procaspasa-9 y luego se autoactivan. El complejo final entonces escinde y activa otras caspasas tales como caspasa-3 que alternadamente escinde posteriormente moléculas importantes en la célula, causando la condensación de la cromatina, la fragmentación de la DNA y conduciendo finalmente a la apoptosis . La figura 2 muestra la vía de activación de la caspasa inducida por el citocromo C.
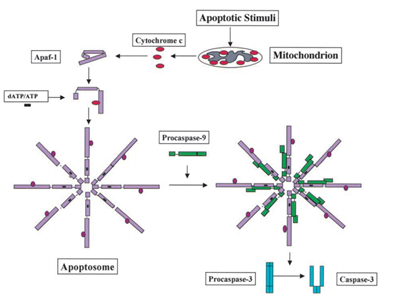
Mihara M et al. los científicos encontraron que el gen p53 de tipo salvaje se puede translocar rápidamente a la superficie mitocondrial de las células tumorales. En el experimento, encontraron que alguna proteína p53 de tipo silvestre inducida por estrés tiene la capacidad de translocarse a las mitocondrias de timocitos en células humanas o de ratón después de la apoptosis debido al daño del ADN y la hipoxia. Luego, estas proteínas p53 de tipo salvaje inducen la permeabilización de las mitocondrias y causan una serie de cambios que se producen en las mitocondrias como la liberación del citocromo c mediante la formación de complejos con Bcl2 y BclXL .,
como un buen resultado clínico con poco efecto secundario, la terapia génica es popular. A finales de 2005, había 1020 ensayos de terapia génica en la base de datos de Journal of Gene Medicine. Entre estos ensayos, 66% de las terapias génicas se llevaron a cabo en pacientes con cáncer, y en 58 ensayos se utilizó rAd-p53, un adenovirus recombinante que codifica el gen p53 humano. En abril de 2004, se lanzó formalmente una inyección recombinante de adenovirus humano-p53 (Gendicine). Gendicine se usa para tratar el carcinoma de células escamosas de cabeza y cuello y fue aprobado por la Administración Estatal de alimentos y Medicamentos de China en octubre. 16, 2003., Se convirtió en el primer producto de terapia génica en el mundo en ser aprobado por el gobierno chino .
el gen p53 fue descubierto para regular el metabolismo en 2005. Para transferir de la fase G1 a la fase S, Las células deben tener suficiente soporte de materias primas para la síntesis de ADN, orgánulos y proteínas. Para regular este proceso, son necesarios algunos puntos de control. Uno de ellos es el punto de control dependiente de glucosa en G1/S. está regulado por la proteína quinasa activada por AMP (AMPK). Cuando se agota la glucosa, la AMPK puede fosforilar la proteína p53, que a su vez induce la detención celular y evita la muerte celular., Las células que se encuentran con la detención dependiente de p53 volverán a entrar en el ciclo celular cuando la glucosa es suficiente .
se sabe que la inactivación de p53 es necesaria para la formación de tumores. Bykov et al. VJ y Snydel El et al. señale que el funcionamiento incorrecto de p53 puede conducir a la proliferación de un tumor existente . Ventura y sus compañeros de trabajo hicieron algunos experimentos para probar esta hipótesis. Restauraron la función del p53 endógeno en tumores autóctonos primarios para examinar la consecuencia de la reactivación del p53., El resultado mostró que la reactivación de p53 fue responsable de la regresión de tumores autóctonos. Eso significa que la proteína p53 inactivada puede conducir al desarrollo del tumor . Xue y otros científicos también hicieron un experimento para probar la consecuencia de la reactivación de p53 en tumores. Utilizaron la interferencia reversible del ARN (RNAi) para regular la expresión de p53 endógeno en ratones con cáncer de hígado. En el experimento, la doxiciclina (Dox) se usa para reactivar p53, ya que la expresión de p53 se suprime totalmente cuando falta Dox y se restaura rápidamente cuando se agrega Dox., Cuando se trató con Dox, el miARN p53 se apagó, lo que a su vez provoca un aumento de la expresión de p53. El resultado mostró que los tumores en ratones tratados con Dox se vuelven indetectables después de 12 días, mientras que los tumores en ratones no tratados crecieron rápidamente. Para probar la consecuencia de la reactivación transitoria de p53, trataron ratones con Dox durante 4 días y luego se detuvieron. El resultado mostró que incluso un tratamiento de dos días puede causar regresión de los tumores y 4 días de tratamiento pueden causar una regresión completa de los tumores., También señalaron que durante la regresión tumoral, la reactivación transitoria de p53 puede desencadenar la senescencia celular, no la apoptosis. En el mismo año, Hu encontró que la implantación embrionaria en ratones hembra p53 -/ – está regulada por el factor inhibidor de la leucemia (LIF). La LIF es una citocina secretada y es importante para la implantación de blastocitos. El gen codificador LIF se identifica como el gen objetivo p53 y el sitio de unión p53 se encuentra en intrón 1 tanto en humanos como en ratones .