Introduction
la mortalité des porcelets est l’un des principaux traits de sélection dans l’élevage porcin et est influencée par la truie, les porcelets et l’environnement. Par conséquent, la mortalité des porcelets est un phénotype complexe et dépend de la capacité de la truie à élever sa progéniture, mais est également fonction du poids à la naissance, de la gestion et de la sélection (Knol et al., 2002)., Cependant, les défauts récessifs monogéniques contribuent également à la mortalité des porcelets, bien que seuls quelques exemples aient été rapportés dans le passé (Murgiano et al., 2012; Matika et coll., 2019). Même dans les cas où l’effet de la mutation est sévère, la sélection efficace contre une telle mutation est entravée par la basse fréquence. Dans de nombreux défauts graves, les zygotes meurent très tôt dans la gestation, ne laissant aucune trace autre que l’absence d’homozygotes dans la population en général (Derks et al., 2019).,
Les effets de consanguinité dans les populations de porcs commerciaux sont généralement maîtrisés par la sélection sélective pour réduire la mortalité chez les porcelets en améliorant à la fois les capacités maternelles et la viabilité des porcelets (Olijslagers, 2018). Cependant, les variants sous-jacents aux défauts monogéniques récessifs ne sont pas bien capturés dans les valeurs de reproduction et peuvent dériver vers des fréquences plus élevées à la suite d’une sélection intense (Georges et al., 2019). De plus, ces variantes peuvent également être maintenues à la suite d’une sélection d’équilibrage pour un effet positif corrélé à l’état hétérozygote (Derks et al., 2018).,
Les défauts récessifs ne contribuent que marginalement à la mortalité globale des porcelets (Alonso-Spilsbury et al., 2007). Néanmoins, les variants affectant la mortalité des porcelets sont d’une grande importance car ces variants influencent directement la production et le bien-être animal (Baxter et al., 2013; Rutherford et coll., 2013). Cependant, dans la gestion des populations animales, l’apparition de défauts à basse fréquence est généralement mal documentée (souvent des termes très généraux sont utilisés), et les syndromes ne sont souvent reconnus qu’une fois qu’ils ont atteint une fréquence élevée., Ceci est particulièrement pertinent pour les syndromes qui ne conduisent pas à des phénotypes très distincts. Par conséquent, même dans les populations reproductrices commerciales, peu de suivi peut être fait sur des syndromes spécifiques, et pour sélectionner efficacement contre des syndromes de basse fréquence spécifiques, il faut donc de nouvelles approches.
dans ce travail, nous décrivons la découverte d’un syndrome hautement débilitant dans une population porcine commerciale à travers une enquête basée sur un réseau SNP de densité moyenne combiné et le séquençage du génome entier (WGS)., L’étude a mené à l’identification d’une délétion frameshift de 16 PB dans le gène SPTBN4, avec des conséquences phénotypiques claires prédites chez les homozygotes. La fréquence porteuse est d’environ 9% dans la population étudiée, affectant environ 0,81% des portées de la population. La fréquence était suffisamment faible pour être inconnue pour avoir une base génétique, et même être effectivement non reconnu comme un syndrome spécifique du tout. Lors de la mise en œuvre de l’enquête, une truie gravide a été identifiée par un sanglier porteur., Les porcelets affectés souffrent de myopathie et sont incapables de marcher, entraînant généralement la mort quelques heures après la naissance, tout à fait en ligne avec la pathologie prévue par rapport à des cas humains et souris similaires.
matériel et méthodes
Animaux, génotypes et prétraitement
l’ensemble de données comprend 31 839 animaux issus d’une lignée de sanglier synthétique à grand fond blanc. La lignée est maintenue et élevée dans les fermes Topigs norsvin nucleus, principalement en sélectionnant des traits de production et de santé., Les animaux ont été génotypés sur la puce SNP Illumina GeneSeek custom 50K (Lincoln, NE, USA). Les animaux avec une fréquence de génotypes manquants > 0,15 ont été enlevés. Nous avons écarté les marqueurs qui ne répondaient pas aux critères de filtrage suivants: un taux d’appel minimum de 0,85, une fréquence d’allèle mineur > 0,01 et une valeur de test exacte de Hardy-Weinberg en dessous de P < 10-12. De plus, les marqueurs dont l’emplacement est inconnu sur la construction du génome Sscrofa11.1 ont été écartés, laissant 41 573 marqueurs après filtrage., Toutes les étapes ont été effectuées dans Plink v1.90b3 .30 (Purcell et al., 2007).
phasage de L’Haplotype et Identification de L’Haplotype SSC6
Nous avons effectué le phasage de l’haplotype et l’imputation des sites manquants dans Beagle5.0 avec le paramètre pour la taille effective de la population fixé à 100, d’autres paramètres étaient par défaut (Browning et al., 2018). Les homozygotes attendus ont été estimés sur la base de la fréquence des haplotypes, en utilisant le principe de Hardy-Weinberg. Un test binomial exact a été appliqué pour tester le nombre d’homozygotes observés avec le nombre d’homozygotes attendus., L’haplotype a été considéré comme significativement appauvri si P < 5 × 10-3.
effets phénotypiques associés à L’Haplotype SSC6
Nous avons examiné l’haplotype SSC6 pour les enregistrements sur le nombre total de porcelets nés, mort-nés, momifiés, la survie de mise bas et la survie en lactation (survie jusqu’à environ 21 jours) d’un total de 9 666 portées. Nous avons répertorié ces phénotypes pour toutes les portées de CXC et de CxN identifiées. Nous avons utilisé un test T de Welch pour évaluer si les phénotypes des portées CxC diffèrent significativement des portées CxN. Une valeur p < 0.,05 a été considéré comme significatif.
analyse du séquençage du génome entier et Identification des variantes candidates
l’ensemble de données comprend 71 individus séquencés du génome entier de la population étudiée. Les 71 échantillons étaient également présents dans notre ensemble de données de 31 839 animaux génotypés sur le 50K. les 71 échantillons ont un volume total de 1,93 Tbp (paires de bases tera) à partir de 14,16 milliards de lectures d’extrémité appariée de 150 bp (tableau S3). Les échantillons ont été séquencés sur Illumina HiSeq 2000. Nous avons aligné les séquences sur la construction du génome Sscrofa11.1 en utilisant BWA-mem version 0.7.,15 (Li et Durbin, 2009) avec une mappabilité moyenne de 98,9% et une couverture d’échantillon allant de 8,8 à 14,8 X (moyenne de 10,9 X). Samblaster a été utilisé pour supprimer les doublons PCR (Faust et Hall, 2014). Samtools a été utilisé pour trier, fusionner et indexer des fichiers bam (Li et al., 2009). La cartographie et les statistiques de qualité ont été générées à L’aide de Qualimap (Okonechnikov et al., 2016). L’appel Variant a été effectué avec Freebayes v1.1. 0 avec les paramètres suivants: –min-base-quality 10 –min-alternate-fraction 0.2 –haplotype-length 0 –min-alternate-count 2 (Garrison et Marth, 2012)., Les variantes avec un score de qualité Phred < 20 ont été rejetées (Li et al., 2009). Les variantes ont été annotées à l’aide du prédicteur D’effet variant Ensembl (VEP, release 96) (Mclaren et al., 2016). L’impact des variants missense a été prédit en utilisant le tri intolérant de tolérant (SIFT) (Kumar et al., 2009). L’analyse LD a été réalisée à L’aide de Plink v1. 90b3.30 (Purcell et al., 2007) avec les paramètres suivants –Chr-set 18, –r2, LD-window-R2 0.8.
alignement des protéines SPTBN4
l’alignement des protéines entre le type sauvage et la protéine mutante a été effectué à L’aide de ClustalO (Madeira et al.,, 2019) et visualisé à L’aide D’ESPript 3 (Robert et Gouet, 2014). Une visualisation et une validation supplémentaires ont été effectuées à l’aide de JBrowse genome viewer version 1.12.1 (Skinner et al., 2009).
Validation de la délétion causale Sptbn4 de 16 bp
la PCR a été réalisée en utilisant 60 ng d’ADN génomique, avec 0,4 µm de chaque amorce, 1,8 mM MgCl2 et 25 unités / ml D’ADN polymérase Onetaq® (mélange maître Onetaq® 2x avec tampon Standard, New England Biolabs) dans le tampon PCR du fabricant dans un volume final de 12 µl., La dénaturation initiale pendant 1 min à 95°C a été suivie de 35 cycles de 95°C pendant 30 s, 55°C pendant 45 s, 72°C 90 s, suivis d’une extension de 5 min à 72°C. Les amorces de PCR pour SPTBN4 sont TCAAGGGTGCAGGCTCTCTTC forward et ggtaggaagctcgaagtggg reverse. L’amorce avant a été marquée par colorant avec L’un ou l’autre 6-FAM pour produire un produit de PCR marqué par fluorescence détectable sur le séquenceur D’ADN ABI 3730 (Applied Biosystems). La taille des fragments a été déterminée à l’aide du logiciel GeneMapper 5 D’ABI.,
examen histopathologique
deux porcelets atteints âgés de moins de 1 semaine ont été envoyés au département de pathologie de la santé animale Royale (Deventer) pour examen. Macroscopiquement, toutes les observations étaient dans les limites normales. Les muscles squelettiques de la patte antérieure, du muscle dorsal et de la patte postérieure des deux animaux ont été échantillonnés pour la coloration de routine H&E et PTAH. Le tissu musculaire a été stocké dans des bocaux séparés et fixé dans une solution de formaldéhyde à 4%, tamponnée (=solution de formol à 10%, tamponnée)., Après cela, le tissu a été incorporé dans de la paraffine et tranché en 2 µm selon la procédure d’opération standard (SOP RAH). Par la suite, les lames ont été deparaffinisées et régulièrement colorées pour l’hématoxyline et l’éosine (h&E) dans une machine de couleur automatique. Simultanément, des lames supplémentaires de 2 µm du tissu musculaire ainsi qu’une lame de contrôle positif du tissu musculaire ont été préparées pour la coloration manuelle avec « l’hématoxyline d’acide phosphotungstique”, en abrégé PTAH. Cette coloration est préférée pour démontrer les striations croisées du muscle squelettique.,
valeurs de sélection et analyse des associations
dans cette étude, nous avons évalué 63 caractères utilisés dans le programme de sélection. Les valeurs de sélection estimées déréglées (VEB) ont été utilisées comme variable de réponse pour chaque trait étudié. La valeur de reproduction estimée (VEB) de tous les caractères évalués a été déréglée à l’aide de la méthodologie décrite par Garrick et al. (2009). L’EBV de chaque animal a été obtenu à partir de l’évaluation génétique de routine par un programme de sélection commerciale (Topigs Norsvin) à l’aide d’un modèle animal., La fiabilité par animal aux fins de la dérégression a été extraite de l’évaluation génétique basée sur la méthodologie de Tier et Meyer (2004). Les héritabilités utilisées pour la dérégression ont également été extraites de l’évaluation génétique de routine. Enfin, des facteurs de pondération basés sur la fiabilité estimée de la DEBV ont également été estimés selon Garrick et al. (2009) en utilisant une valeur de 0,5 pour le scalaire C. pour assurer la qualité du DEBV, seuls les animaux avec un facteur de pondération supérieur à zéro et une fiabilité du DEBV supérieure à 0. ,20 ont été utilisés dans les analyses d’association. La fiabilité du DEBV a également été obtenue selon Garrick et al. (2009).
des analyses D’Association ont été effectuées à l’aide du logiciel ASREML (Gilmour et al.,, 2009) en appliquant le modèle animal mixte linéaire suivant:
où DEBVij est la DEBV observée pour l’animal j, w est le facteur de pondération pour le résidu, µ est la moyenne globale de la DEBV de la population, Ri est le statut de Porteur (nombre d’allèles nuisibles) de la mutation 4 i, aj est l’effet génétique additif estimé à l’aide d’une matrice de relation moyenne basée sur le pedigree, et l’erreur résiduelle. Les Associations avec A-log10 (valeur P) supérieure à cinq ont été déclarées comme significatives.
Résultats
1.,Le Segment de 5 Mb sur le Chromosome 6 affecte la survie en Lactation chez les porcs
Nous avons analysé 31 638 animaux d’une seule lignée de sanglier de race pure (lignée synthétique à grand fond blanc), génotypée sur la puce SNP Porcine 50k (construction Sscrofa11.1) (Warr et al., 2019). L’analyse a révélé un segment de 1,5 Mb sur le chromosome 6 (SSC6:48,75–50,25) montrant un déficit d’homozygotie associé à une réduction de la survie en lactation (tableaux 1 et 2). L’haplotype se sépare à une fréquence d’allèles modérée de 4,5% (fréquence porteuse de 9,0%) dans la population étudiée., La fréquence des haplotypes a fluctué au cours de la dernière décennie, mais a diminué au cours des 3 dernières années (Figure S1). Nous avons testé si la fréquence était motivée par un effet d’avantage hétérozygote. Cependant, nous avons trouvé des associations principalement négatives avec des traits de sélection importants, sauf pour la profondeur de la longe et la longueur de gestation (Tableau 3), ce qui suggère que la fréquence est purement le résultat de la dérive génétique.

tableau 1 Caractéristiques de L’haplotype SSC6.,

Tableau 2 les portées porteuses par porteuses montrent une diminution de 24% de la survie en lactation par rapport aux portées porteuses par non porteuses. Les résultats significatifs sont indiqués en gras.

le Tableau 3 Traits associés de façon significative avec les porteurs hétérozygotes de la SPTBN4 suppression.
Les 52 portées porteuses par porteuses (CxC) ne montrent aucune réduction significative du nombre total d’animaux nés ou nés en vie., Cependant, la survie en lactation est réduite d’environ 24% chez les portées CxC par rapport aux accouplements porteurs par non porteurs (CXN), ce qui indique que les porcelets homozygotes meurent pendant la période de lactation (Tableau 2). Ensuite, nous avons examiné les remarques concernant le temps et la cause de mortalité des portées de CXC. Cela a révélé que la plupart des porcelets qui sont morts dans les 24 premières heures après la naissance. La majorité de ces porcelets ont été principalement décrits par les agriculteurs comme « porcelet faible à la naissance.,”
L’analyse du séquençage du génome entier révèle une délétion Frameshift de 16 PB dans SPTBN4 en tant que variante causale probable
pour identifier la mutation causale, nous avons examiné les données de séquence du génome entier de 71 animaux de la population étudiée et identifié cinq animaux porteurs. L’analyse du déséquilibre de liaison (LD) a révélé 267 variants SNP et indel dans un LD élevé (r2 > 0,8) avec L’haplotype SSC6 (tableau S1), la majorité étant dans un LD parfait (247 variants)., Seules cinq variantes affectent potentiellement la séquence de codage (trois missense, un frameshift, un Splice-accepteur). Les trois variants missense sont prédits pour être tolérés par SIFT (score > 0.18, tableau S1), tandis que la variante Splice-accepteur affecte un gène codant un peptide de 28 bp de fonction inconnue, peu susceptible d’être causal. Cependant, une variante de LD complète (r2 = 1) avec l’haplotype a été prédit pour avoir un impact élevé; une délétion frameshift de 16 bp dans l’exon 26 du gène SPTBN4 (6:g. 48801280delgacggtgtacgccggt) (Figures 1A, B)., La délétion frameshift (ENSSSCP00000031537:P. Arg1902fs) introduit 30 nouveaux acides aminés et un codon d’arrêt prématuré, produisant une protéine spectrine bêta Non érythrocytaire 4 altérée et tronquée (SPTBN4). Les Mutants n’ont pas les 662 derniers acides aminés de la protéine de type sauvage( Figure 1C), y compris le domaine d’homologie de pleckstrine (PH) requis pour le transport des protéines vers les membranes (Wang et al., 2018). La protéine SPTBN4 est un membre des protéines bêta-spectrines et est une actine qui relie la membrane cellulaire au cytosquelette d’actine., Les mutations SPTBN4 perturbent la machinerie cytosquelettique contrôlant la bonne localisation des canaux ioniques dans les nerfs myélinisés provoquant des neuropathies motrices (Parkinson et al., 2001; Wang et coll., 2018).

la Figure 1 (A) SPTBN4 gène modèle. L’emplacement du 26ème exon affecté est indiqué en rouge. B) Illustration de la suppression de 16 bp. La Figure montre le type sauvage et l’exon mutant. (C) alignement de la séquence protéique sptbn4 de type mutant (Mt) et sauvage (Wt). La mutation induit 30 nouveaux acides aminés et un codon d’arrêt prématuré.,
le génotypage de cinq portées de CXC confirme la délétion de SPTBN4 comme étant le coupable probable
Nous avons génotypé cinq portées de CxC pour la délétion de 16 PB qui avait au moins deux porcelets (gamme 2-6) qui sont morts dans les 48 premières heures après la naissance. Les cinq portées ont produit 53 porcelets dont 19 homozygotes pour la délétion de 16 PB (Tableau 4). Les 19 porcelets homozygotes sont morts dans les 48 h après la naissance (18 dans les 24 h). Sur les 34 porcelets restants (8 de type sauvage et 26 porteurs), seulement 1 est mort en 48 h, probablement causé par d’autres facteurs (environnementaux).,

Tableau 4 génotypage de la suppression causale probable du frameshift SPTBN4 de 16 bp dans cinq portées porteuses par porteuses. La somme par classe de génotype est indiquée en gras.
les porcelets homozygotes pour la délétion SPTBN4 souffrent de myopathie et de paralysie des membres postérieurs
Nous avons surveillé une portée CXC récente (date de mise bas: 28 avril 2019) qui a produit six porcelets sains, deux touchés (échantillons: 9912, 9916) (Figure 2a) et trois porcelets mort-nés., Nous avons confirmé le statut de délétion de SPTBN4 homozygote pour les deux porcelets touchés (tableau S2). De plus, nous avons observé quatre porteurs hétérozygotes et deux porcelets homozygotes de type sauvage parmi les individus sains. L’un des porcelets mort-nés (échantillon: 9921) était également homozygote pour la délétion, tandis que les deux autres étaient hétérozygotes. Les porcelets atteints souffrent d’une faiblesse musculaire extrême (Figures 2B, C), d’une paralysie des membres postérieurs et de tremblements (vidéo S1). Par conséquent, les porcelets étaient incapables de marcher ou de boire.,
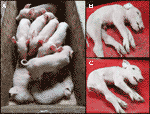
Figure 2 (a) deux porcelets atteints (vivants) ainsi que six litières saines. Les porcelets proviennent d’un accouplement CxC mis bas le 28 avril 2019. B) porcelet mâle touché 9912. C) porcelet femelle touché 9916.
les porcelets atteints manquent de stries croisées dans les Muscles squelettiques des membres dorsaux et postérieurs
l’examen histopathologique a révélé une dégénérescence dispersée des fibres musculaires chez les deux porcelets, ainsi qu’une nécrose focale et une vascularite dans le muscle dorsal chez l’un des porcelets (ID = 9912)., De plus, la coloration à l’hématoxyline à l’acide phosphotungstique (PTAH) montre une coloration divergente des fibres musculaires squelettiques, indiquant une diminution des stries croisées, en particulier dans les muscles des pattes dorsales et postérieures des animaux atteints (Figure 3B), tandis que les pattes avant ne semblent pas affectées (Figure 3A). La diminution des stries croisées est indiquée par une coloration anormale et une perte générale de volume des fibres musculaires (Figure 3B). Les changements histopathologiques observés dans les pattes arrière et dans les muscles dorsaux sont indicatifs de la dystrophie musculaire.,

la Figure 3 (A) vue en coupe Transversale d’un muscle squelettique de la jambe avant. La flèche noire indique la coloration normale (sombre) des fibres musculaires indiquant la présence de stries croisées. Barre de PTAH = 50 µm. B) vue en coupe transversale d & apos; un muscle squelettique depuis la patte arrière. La flèche noire indique une coloration anormale (rose) des fibres musculaires indiquant l’absence de stries croisées. La flèche jaune indique la coloration normale et la présence de stries transversales. Barre de PTAH = 50 µm.,
Discussion
dans ce travail, nous signalons une nouvelle malformation congénitale causant la mortalité des porcelets probablement due à une délétion frameshift de 16 PB dans le gène SPTBN4. Les porcelets souffrent d’une faiblesse musculaire extrême (myopathie) et meurent quelques heures après la naissance. La délétion devrait conférer une perte complète de fonction de la protéine spectrine bêta, Non érythrocytaire 4. SPTBN4 est un membre de la famille des gènes spectrines et est nécessaire pour le regroupement des canaux ioniques aux nœuds de Ranvier, affectant le potentiel d’action (Devaux, 2010)., Les Mutations perturbent la machinerie cytosquelettique qui contrôle la localisation correcte des canaux ioniques et la fonction des domaines axonaux principalement au niveau des segments initiaux de l’axone (AIS) et des nœuds de Ranvier (Wang et al., 2018). Plus précisément, le domaine C-terminal affecté de SPTBN4 est crucial pour le trafic de canaux KCNQ2 et l’excitabilité aux nœuds de Ranvier (Devaux, 2010).
des recherches de suivi ultérieures ont identifié des cas humains et murins qui indiquaient que le syndrome qui s’ensuivrait ne s’avérerait probablement pas immédiatement mortel, mais conférerait plutôt une myopathie sévère., Les données de génotype SNP de densité moyenne, disponibles pour tous les animaux de la population reproductrice (N = 31 839), ont permis d’identifier les porteurs. Parmi ces porteurs, il y avait une truie qui était approximativement à mi-parcours de la grossesse au moment de l’identification, engendrée par un sanglier qui était également porteur. La ferme d’élevage a été avisée de documenter la portée à la naissance. Le phénotype observé des porcelets atteints (myopathie, paralysie des membres postérieurs, tremblements) était tout à fait conforme à ce qui a été observé chez les patients humains présentant une perte de fonction homozygote ou des mutations hétérozygotes composées dans le gène SPTBN4 (OMIM: 606214)., Deux des patients humains ont des mutations de perte de fonction dans le domaine PH (Wang et al., 2018), soutenant qu’une perte du domaine PH chez les porcs entraînerait probablement une perte complète de fonction de la protéine SPTBN4. Chez l’homme, des mutations similaires entraînent une myopathie congénitale sévère causée par l’absence de fibres musculaires de type I, une neuropathie et une surdité (Knierim et al., 2017; Wang et coll., 2018). Wang et coll. (2018) ont également observé une neuropathie axonale motrice chez plusieurs patients caractérisée par une hypotonie congénitale, une faiblesse profonde et une perte de réflexes tendineux profonds dès la petite enfance., De plus, les biopsies nerveuses ont révélé des canaux Na+ nodaux réduits et aucun canal kcnq2 K+ nodal, révélant la pathologie moléculaire à l’origine d’un dysfonctionnement du système nerveux. Par conséquent, nous concluons que cette variante frameshift est la mutation causale probable menant au phénotype observé et à l’épuisement du génotype homozygote dans la population. Des études futures pourraient se concentrer sur la réalisation d’un knock-out in vivo du gène SPTBN4 chez le porc, pour étudier le syndrome et le phénotype associé plus en détail.,
Nous n’avons pas observé de dégénérescence des fibres musculaires dans les pattes avant, alors que les fibres musculaires dorsales et postérieures étaient clairement affectées. Cette observation pourrait expliquer en partie la paralysie des membres postérieurs, alors que les pattes avant ne sont pas affectées. L’écart entre les fibres musculaires des jambes avant et arrière a également été décrit chez des souris frémissantes, chez lesquelles les mutations de la perte de fonction SPTBN4 provoquent une neuropathie motrice, une paralysie des membres postérieurs, des tremblements et une surdité centrale (Parkinson et al., 2001; Komada et Soriano, 2002). Maladie de Parkinson et coll., (2001) décrivent des vitesses de conduction nerveuse réduites dans les nerfs sciatiques de souris avec des allèles frémissants provoquant la neuropathie périphérique du membre postérieur. L’Expression de SPTBN4 chez la souris est limitée au cerveau, à la moelle épinière et aux nerfs sciatiques et n’est pas observée dans le muscle squelettique, de sorte que cette maladie est principalement un défaut neuronal. Dans l’ensemble, on ne sait pas quel mécanisme provoque l’absence de symptômes dans les membres antérieurs. Ce « knock-out naturel » chez les porcs peut être une ressource utile pour étudier la maladie humaine, car les porcs sont généralement un meilleur modèle pour étudier la maladie humaine par rapport aux espèces de rongeurs., De plus, la conséquence de la perte de la fonction SPTBN4 peut être étudiée plus en détail.
la taille effective de la population (Ne) de la race étudiée est estimée à environ 100 (Hidalgo et al., 2016). En élevage, un faible Ne augmente le risque que les allèles nuisibles augmentent en fréquence par hasard. De plus, des études antérieures ont montré que les allèles létaux récessifs peuvent être entraînés par des effets avantageux chez les hétérozygotes (Derks et al., 2018; Matika et coll., 2019). Matika et coll.,, 2019 a trouvé une mutation récessive acquise par arrêt dans le gène MSTN associée à une augmentation majeure de la profondeur musculaire chez les hétérozygotes. Cependant, nous ne trouvons aucune preuve d’un avantage hétérozygote dans notre étude. Avec les techniques génomiques actuelles, nous pouvons maintenant identifier les allèles délétères dérivant vers des fréquences plus élevées et surveiller l’émergence de nouveaux allèles délétères avec précision, ce qui permet une purge plus efficace. De plus, le résultat de ce type d’étude améliorera considérablement la conscience des défauts génétiques « cachés” au niveau de l’éleveur et de l’agriculteur., Sans aucune information préalable, les malformations congénitales rares sont souvent enregistrées comme « porcelet faible. »Et sans autre distinction de syndromes spécifiques, une action supplémentaire n’est pas possible. Dans la plupart des cas, on ne sait pas s’il existe une base génétique ou s’il peut y avoir d’autres effets confondants. Avec des informations génomiques antérieures, le syndrome peut être identifié, comparé à d’autres cas, et les porteurs identifiés, conduisant à des informations exploitables.
la mortalité des porcelets est d’une grande importance économique et pour le bien-être des animaux., Par conséquent, la découverte de la mutation SPTBN4 a conduit à une mise en œuvre immédiate dans le programme de sélection pour minimiser la fréquence des accouplements transporteur par transporteur. Cela permet d’éviter la naissance d’individus affectés, améliorant ainsi le bien-être animal et réduisant les pertes économiques.
Conclusion
dans cette étude, nous signalons une nouvelle anomalie congénitale probablement causée par une délétion récessive du gène SPTBN4 chez le porc. Les résultats sont étayés par des similitudes frappantes avec les phénotypes syndromiques associés à SPTBN4 chez l’homme et la souris., L’étude permet de surveiller et de purger l’allèle délétère de la population. Les croisements transporteur par transporteur peuvent être évités, excluant les individus affectés, réduisant ainsi les pertes économiques et améliorant le bien-être des animaux. Enfin, ces” knockouts naturels » obtenus dans l’industrie de la sélection peuvent fournir un modèle pour la maladie humaine et améliorer notre compréhension de la fonction des gènes au sein du clade des mammifères, et fournir un modèle potentiel pour la maladie humaine.,
énoncé de disponibilité des données
énoncé D’éthique
l’examen éthique et l’approbation N’étaient pas nécessaires pour l’étude chez l’animal parce que les données utilisées dans cette étude ont été obtenues dans le cadre de la collecte de données de routine des programmes D’élevage Topigs Norsvin, et non spécifiquement aux fins de ce projet. Par conséquent, l’approbation d’un comité d’éthique n’était pas obligatoire. La collecte des échantillons et l’enregistrement des données ont été effectués strictement conformément à la loi néerlandaise sur la protection et le bien – être des animaux (Gezondheids-en welzijnswet voor dieren)., Le consentement éclairé des propriétaires a été obtenu par écrit pour la participation de leurs animaux à cette étude.
contributions des auteurs
MG, H-JM et MD ont conçu et conçu l’étude. BH était responsable de l’organisation générale et de la communication avec Topigs Norsvin et les agriculteurs. MD et ML ont effectué l’analyse des données. BD et KL ont effectué des travaux de laboratoire. SG-V a effectué l’analyse pathologique. MD a écrit le manuscrit. H-JM, MG, BH, SG-V, BD, KL et ML ont fourni des commentaires et des suggestions utiles et ont aidé à rédiger le manuscrit. Les données phénotypiques ont été analysées par ML., Tous les auteurs ont lu et approuvé le manuscrit final.
Financement
cette recherche a été financée par le partenariat STW-Breed4Food, numéro de projet 14283: From sequence to phenotype: detecting déleterous variation by prediction of functionality. Cette étude a été soutenue financièrement par NWO-TTW et les partenaires de Breed4Food Cobb Europe, CRV, Hendrix Genetics et Topigs Norsvin. De plus, cette étude a été soutenue par le projet IMAGE (Horizon 2020, No.677353). Les bailleurs de fonds n’ont joué aucun rôle dans la conception de l’étude, la collecte et l’analyse des données, la décision de publier ou la préparation du manuscrit., L’utilisation du cluster HPC a été rendue possible par CATAgroFood (installations de recherche partagées Wageningen UR).
avertissement
Les données utilisées dans cette étude ont été obtenues dans le cadre de la collecte de données de routine des programmes de sélection Topigs Norsvin, et non spécifiquement aux fins de ce projet. Par conséquent, l’approbation d’un comité d’éthique n’était pas obligatoire. La collecte des échantillons et l’enregistrement des données ont été effectués strictement conformément à la loi néerlandaise sur la protection et le bien – être des animaux (Gezondheids-en welzijnswet voor dieren).,
conflit d’intérêts
ML et BH sont des employés du Centre de recherche Topigs Norsvin, un institut de recherche étroitement lié à L’un des bailleurs de fonds (Topigs Norsvin).
Tous les autres auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière pouvant être interprétée comme un conflit d’intérêts potentiel.
Les autres Partenaires de Breed4Food Cobb Europe, CRV, Hendrix Genetics, déclarent n’avoir aucun intérêt concurrent pour cette étude.,
Remerciements
Les auteurs remercient Frank van Haaren et Toon Janssen d’avoir organisé la communication avec la ferme. Nous remercions Manon Houben pour sa contribution aux examens pathologiques. Nous remercions Mout Viller d’avoir pris des vidéos et des photos des porcelets touchés et en bonne santé après la naissance. Nous remercions Jürgen Harlizius d’avoir pris soin des porcelets après la remise. Nous remercions Gerda van Eldik pour la collecte des échantillons. Nous remercions Egbert Knol et Egiel Hanenberg pour le soutien général et les conseils de Topigs-Norsvin.,
matériel supplémentaire
Vidéo S1 / vidéo montrant les deux personnes touchées après la naissance.
tableau S1 / variation génomique de la DL élevée avec L’haplotype SSC6.
tableau S2 / génotypage de la portée CXC incluant deux individus atteints (date de mise bas: 28 avril 2019). Le tableau montre le statut del/del homozygote pour les deux porcelets touchés 9912,9916.
tableau S3 / cartographie et statistiques de couverture à partir d’échantillons WGS.
Figure S1 / SPTBN4 fréquence porteuse de suppression de 2007-2019.