The gene p53 was first discovered in 1979. Uma proteína foi identificada nas células do Ratinho transformadas em 40 pelo vírus símio (SV40) por imunoprecipitação com soro anti-T; esta proteína foi chamada de proteína p53 . No mesmo ano, Kress e outros cientistas encontraram uma nova classe de proteínas com uma massa molecular variando de 50-60kDa. Este tipo de proteína foi identificado como p53 . A proteína p53 também pode ser identificada a partir de várias linhas celulares transformadas por imunoprecipitação., Lane e Linzer também tiveram um resultado semelhante em 1979. Outras evidências para identificar o p53 é que o p53 foi expresso em todas as células testadas de ratos transformados; estes testes incluem sarcomas quimicamente induzidos, fibroblastos transformados e leucemias, enquanto nas células normais, o p53 não foi expresso. Além disso, um alto nível de p53 foi detectado na maioria das células transformadas, não importa como as células foram transformadas, quer espontaneamente ou não espontaneamente ., Isso foi em grande parte devido ao aumento da estabilidade do p53, no entanto, em células de carcinoa embrionária F9, ele expressou um alto nível de p53, isso foi devido à quantidade de RNAm traduzida p53 .
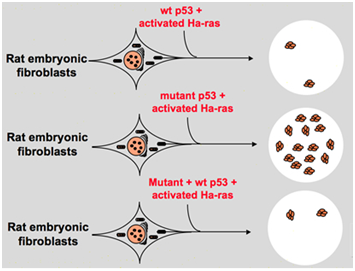
Após a proteína p53 ter sido descoberta em 1979, tornou-se popular para analisá-la. No entanto, naquela época, como era uma proteína recém-descoberta, e não havia um nome anterior para ela, diferentes instituições usaram nomes diferentes e artigos publicados com nomes diferentes., A fim de resolver este problema, em 1983, durante o 1º workshop Internacional p53 realizado em Oxted, Reino Unido, cientistas de diferentes grupos de pesquisa em diferentes países se reuniram para discutir uma nomenclatura comum para esta proteína recém-descoberta. Neste encontro,” p53 ” se torna seu nome e tem sido usado desde então. Pensou-se que a razão pela qual os cientistas chamaram a proteína p53 é que a massa molecular desta proteína é 53kDa que é baseada em sua migração em gel SDS. Mais tarde, provou-se que a massa molecular estava errada, e a massa molecular correcta devia ser de 43.,7kDa porque p53 contém uma região rica em prolina, e esta região pode reduzir a migração de p53 em gel SDS. Mas o nome “p53” permaneceu . durante a década de 1980, acreditava-se que a proteína p53 estava envolvida no ciclo celular, além de desempenhar um papel na replicação do DNA. Mais tarde, entre 1982 e 1994, as pessoas descobriram que algumas oncoproteínas virais eram capazes de se ligar ao p53, formando um complexo. Em 1982, Sarnow et al. verificou-se que o adenovírus E1b (58kDa) pode interagir com uma proteína 54kDa que está presente nas células do rato transformadas em SV40 acima mencionadas., De acordo com os resultados das especificidades imunológicas dos anticorpos T e dos mapas peptídicos da proteína 54kDa, esta proteína 54kDa é identificada como p53 . No mesmo ano, os cientistas descobriram que se injetassem o anticorpo p53 nas células do Ratinho 3T3 Suíço, ele inibiria as células entrando na fase S do ciclo celular, no entanto, sob a mesma situação, o anticorpo p53 não afetou SV40 ou o adenovírus induziu a síntese de DNA .,mais tarde, em 1984, os cientistas examinaram o efeito do p53 sobre os fibroblastos 3T3 não transformados; analisaram a taxa de síntese da proteína p53 em diferentes pontos de tempo e descobriram que no final da fase G1, a taxa de síntese e o nível da proteína p53 e seu aumento de mRNA relacionado. Este resultado sugere que a proteína p53 inibe as células que entram na fase divisória da interfase . Maltzman w et al. fez outra experiência no mesmo ano. Eles trataram a célula do rato não transformada com luz UV e carcinógeno químico UV-mimético 4NQO, e eles detectaram um alto nível de p53., O resultado mostrou que a expressão elevada de p53 não é apenas um símbolo que indica o ciclo celular, mas também mais importante um componente que está envolvido na síntese de DNA e proliferação celular . In 1987, when studying the complex of T antigen of simian virus 40 and DNA polymerase α, Gannon, and other scientists found a similar change in the antigen when bound to p53 and polymerase α. Eles também descobriram que a uma certa concentração dos três componentes, eles podem formar um complexo trimérico especial que inclui antigénio T, p53 e DNA polimerase α., Como o antigénio T está envolvido na replicação do ADN viral e na transformação celular, este resultado indica que o p53 desempenha um papel no controlo do ciclo celular e na replicação do ADN .como o experimento mostrou acima, p53 tem a capacidade de imortalizar células. Em 1984, Eliyahu D et al. descobriu que o p53 e o produto do oncogene myc compartilhavam algumas propriedades similares. Ambos têm a capacidade de se ligar a outras proteínas e estão envolvidos no ciclo celular, e ambos se acumulam em núcleos de células transformadas ., Bienz, Pennica e Oren analisadas as seqüências de aminoácidos da proteína p53 e o produto de myc, e eles descobriram que as duas proteínas mostrar semelhanças na estrutura molecular e a posição de especial carregada de resíduos. Em seguida, os cientistas propuseram que p53 pode agir como um oncogene. Baseado nesta hipótese, Eliyahu D et al. fiz algumas experiências. Como a taxa primária de fibroblastos embrionários pode ser transformada pelo envolvimento tanto do produto myc e Ha-ras, as células renais primárias de ratos bebés também podem ser transformadas pela cooperação de Ha-ras e adenovirus região 1A, Eliyahu D et al., decidiu utilizar este tipo de Sistema de testes biológicos para identificar a função oncogénica do p53. Eles trataram células embrionárias normais com p53 e Ha-ras ativados. O resultado mostrou que as células-alvo encontram alterações morfológicas e produzem níveis elevados de p53, Eliyahu D et al. pensava que a transformação dos fibroblastos embrionários por p53 e Ha-ras explicava que o gene p53 é um oncogeno ., Em 1985, Jenkins propôs que o gene p53 pode estender o tempo de vida das células, melhorar a afetividade de transformação, reorganizando as suas sequências codificantes que poderia causar a produção estável de proteínas .
no entanto, no final dos anos 80, os cientistas começaram a perceber que p53 é um gene supressor de tumor em vez de um oncogeno. Eles observaram que p53 com função normal não pode ser detectado em muitos dos tumores e descobriram que perder a expressão e função do gene p53 tipo selvagem é necessário durante a transformação celular., Estes aumentam a possibilidade de que o gene p53 Selvagem possa inibir a progressão neoplásica . Então eles formularam outra hipótese: o gene clone p53 usado em experimentos anteriores contém mutações negativas dominantes dentro do domínio altamente conservado ocasionalmente, o que leva a resultados de experimentos opostos . Em 1988, Ben e outros cientistas detectaram uma enorme quantidade de p53 rearranjada nas linhas celulares murine erythroleukemia — DP20 – 1 e CB3 que são derivados dos bicos de murine infectados com o vírus de leucemia amigo ., Em 1989, Eliyahu, que apontou que p53 é um oncogene mudou de idéia, e ele supôs que o gene p53 selvagem pode inibir a transformação celular. Eliyahu e outros cientistas estudaram o efeito da proteína p53 de tipo selvagem codificada por plasmídeos e p53 mutante na capacidade de provocar transformação embrionária em fibroblastos de taxa primária por várias combinações de oncogeno in vitro. Por exemplo, o mutante p53 mais ras, e o myc mais ras., O resultado mostrou que o tipo selvagem p53 leva a uma enorme redução dos focos transformados causados pelo mutante p53 mais ras; o mutante p53 não mostrou inibição em focos transformados causados por myc mais ras, enquanto a transformação mediada por myc mais ras é muito sensível à expressão do tipo selvagem p53. A figura 1 mostra esta experiência de forma concisa. Mostrou que, em comparação com o mutante p53, o tipo selvagem p53 exibe um efeito inibitório óbvio na transformação celular. O efeito está positivamente relacionado com o nível de expressão do tipo selvagem p53 e negativamente relacionado com o nível de expressão do mutante p53., Este experimento sugeriu que p53 tipo selvagem pode realmente ter uma função oposta em comparação com p53 mutante e pode inibir a tumorigénese . Atualmente, p53 é reconhecido como um gene supressor de tumor. Estima-se que cerca de metade dos tumores são causados por p53. É um dos genes mais frequentemente mutados em humanos, e o gene mais frequentemente analisado em todo o mundo .
durante os primeiros anos da década de 1980, a via bioquímica de p53 e o efeito da mutação p53 não foram claros. In 1991, Kern and other scientists found that a 33-base pair DNA sequence binds specifically to wild-type p53 in vitro. Eles também descobriram que a proteína p53 contém duas mutações que são geralmente encontradas em tumores humanos que não podem se ligar a esta região específica de DNA. Então eles supuseram que a função de p53 depende de sua capacidade de ligar sequências específicas de DNA, e essa capacidade é alterada por mutações encontradas em tumores humanos., Eles também supõem que esta sequência de DNA de 33 pares de bases pode não ser a única sequência que tem a capacidade de se ligar especificamente ao p53 em humanos; no entanto, pode ajudar as pessoas a entender melhor a função do p53 . Mais tarde, p53 foi encontrado para desempenhar um papel durante o ciclo celular, reparação do DNA, diferenciação, iniciando apoptose e angiogênese. Rotter V et al. descobriu-se que o p53 regula a diferenciação das células. Por exemplo, um alto nível de proteína p53 foi detectado em várias etapas-chave durante a diferenciação das células-B. O p53 elevado também pode ser detectado durante a espermatogénese., Enquanto isso, apenas um nível muito baixo de proteína p53 pode ser detectado em alguns órgãos de ratos adultos .em 1990, uma ferramenta útil foi descoberta ocasionalmente. É um mutante sensível à temperatura de p53, chamado p53val135. Ele pode atuar como um verdadeiro tipo selvagem p53 à temperatura de 32,5 oC, suprimindo a transformação, e também pode atuar como outro mutante p53 à temperatura de 37,5 oC ou acima de 48oC, provocando transformação. Além disso, para as células transformadas que expressam p53val135, sua proliferação é controlada à temperatura permissiva, e este tipo de controle é reversível., Ao utilizar este mutante p53val135, descobriu-se que o tipo selvagem p53 induz paragem do ciclo celular em G1 ou G2/M. Em 1991, Elisheva et al. verificou-se que o p53val135 sensível à temperatura desempenhava uma função diferente na linha celular de leucemia mielóide murina. Após a reativação do p53val135 por alguns dias, todas as células morreram, e esta morte exibe algumas propriedades de apoptose . Um ano depois, um resultado semelhante foi obtido por Shaw. Um tipo selvagem p53 foi transfectado em uma linha celular derivada de tumor no cólon humano EB., As células foram examinadas sob microscópios de luz e elétrons, e descobriu-se que exibem algumas propriedades de apoptose . In 1990, Scheffner et al. e outros cientistas descobriram que o E6 que estimula a destruição das proteínas reguladoras das células hospedeiras é codificado pelo papilomavírus humano oncogênico tipos 16 e 18, e pode formar um complexo com o tipo selvagem p53 in vitro, que por sua vez causa a degradação da proteína p53 .
em 1992, uma proteína chave MDM2 foi descoberta porque se liga firmemente com p53, e inibe a transativação mediada por p53., A massa molecular de MDM2 é de 90kDa, e forma um complexo com mutação e tipo selvagem p53 . No mesmo ano, Livingstone RL et al. estudou-se se a célula perdeu uma ou ambas as cópias dos alelos tipo p53 selvagens e se isso foi suficiente para causar a amplificação genética. A amplificação genética foi detectada principalmente em células transformadas, mas não nos fibroblastos normais. O resultado mostrou que as células que perderam uma cópia dos alelos p53 atuam como tipo selvagem p53, enquanto as células que perderam ambas as cópias dos alelos do tipo selvagem p53 exibem uma maior frequência de amplificação . Outra experiência feita por Yin y et al., mostrou um resultado semelhante .
em 1993, um gene alvo p53 chamado CDKN1A foi identificado. Codifica a proteína p21, que é um inibidor cinase dependente das ciclinas que inibe a cyclin-CDK2 e a CDK1 ligando-se a eles. Em 1993, Szekely descobriu que o antígeno nuclear do vírus Epstein-Barr 5(EBNA-5) é codificado pelo vírus Epstein-Barr, e pode infectar células linfoblastóides humanas. Um peptídeo longo de 66 aminoácidos é responsável pela formação do complexo EBNA-5-p53, mutações pontuais de p53 não afetaram a sua capacidade de ligação ao EBNA-5., No entanto, inibe suas formações de complexos com outras moléculas . Em 1994, Cho e seus colegas de trabalho descreveram pela primeira vez a estrutura cristalina do complexo P53-DNA. Este domínio de ligação de DNA também foi chamado de domínio Central. Contém resíduos 102-292 e é constituído por uma sanduíche beta. Eles também demonstraram a estrutura detalhada do domínio principal . Também em 1994, Wang XW et al. a interacção entre a proteína X do vírus da hepatite B (HBX) e a proteína p53 de tipo selvagem no ser humano., Eles descobriram que o HBX pode inibir a capacidade do p53 de se ligar a outro DNA específico da sequência depois de estar ligado ao p53 e também pode inibir a associação do p53 com fatores de transcrição .
em 1997, Honda R et al. a primeira hipótese foi que MDM2 pode desencadear a ubiquitilação p53 e levar à degradação de p53 por um sistema ubiquitin-proteosoma. Eles apontaram que MDM2 se liga ao Domínio N-terminal (NTD) de p53 e atua como ligase ubiquitina E3 . Também em 1997, duas novas famílias de proteínas, p63 e p73, foram descobertas que compartilham homologia substancial com p53., p73, também chamada proteína do tumor 73, é codificada por um gene localizado em 1p36. A localização é apagada frequentemente em neuroblastoma e outros tumores. p73 pode ativar genes alvo p53 e interage com p53 . Yang et al. descobriu-se que o gene p63 está localizado em 3t27-29 e pode ser detectado em várias células humanas e mouse. Como p73, p63 pode transativar genes alvo p53 significativamente, também pode induzir apoptose. Uma característica do p63 é que a maioria do p63 não tem um n-terminus ., No mesmo ano, Serrano e colegas de trabalho descobriram que os fibroblastos murinos primários podem ser transformados por ras na ausência de p53 ou p16, e inativos p53 ou p16 podem facilitar o processo de imortalização das células humanas. Estes achados sugerem que o p53 desempenha um papel na senescência celular . Então, em 1997, ap53 foi encontrado para desempenhar um papel no início da apoptose. Quando as células entram na fase de proliferação, os telómeros no final de cada cromossoma diminuem após cada ciclo de replicação do ADN devido à replicação incompleta do ADN de uma única cadeia no final do suporte de ADN ., O gene supressor do tumor activado p53 limita o número de vezes que a divisão celular pode ocorrer. Wynford TD descobriu que com a perda da função de tipo selvagem p53, todos os fibroblastos escapam da apoptose. Além disso, a função de transativação de p53 pode ser ligada por apoptose . Wynford TD propôs que existem três possibilidades de como p53 é ativado. O primeiro é a modificação pós-translacional por fosforilação, o segundo é up-regula os cofactores transcritionais como p33ING1, o último é down-regula os inibidores p53 como MDM2 .
em 2000, Brodsky MH et al., estudou os alvos de transcrição do p53 em Drosophila. Há evidências de que os olhos de Drosophila exibem um fenótipo ocular severo sob a expressão de p53 humano que irá induzir apoptose das células do disco imaginário ocular, causando a perda de células pigmentadas, inibindo finalmente o desenvolvimento ocular de Drosophila , então Drosophila pode ser um animal modelo para estudar a função de p53. Brodsky descobriu que o gene rpr contém um local de ligação consensual p53 que está localizado na região regulatória cis de rpr, e também é um ativador de apoptose., Com outras evidências, Brodsky alegou que o rpr é um alvo transcritional do p53 . Em 2001, Derry e colegas de trabalho descobriram que C. elegans não tem um gene p53, mas contém um gene cep – 1 que codifica proteínas que têm uma sequência semelhante com a proteína p53. Este gene C. elegans codifica a proteína CEP – 1 que tem a capacidade de induzir apoptose por estresse genotóxico e é um componente necessário durante a meiose .
em 2002, Tyner e colegas de trabalho propuseram que p53 desempenha um papel na regulação do envelhecimento dos organismos., A fim de estudar a função de p53, eles criaram ratos geneticamente modificados com p53 mutado, apagando exons 1-6 e uma região upstream do gene p53 tipo selvagem( p53+/+), chamado p53 + / m. Ela atua como p53 tipo selvagem e tem aumentado a resistência a tumores espontâneos melhor do que o tipo selvagem p53. No experimento, eles monitoraram os ratos contendo p53+/ m, p53+/+ e p53+/ -. p53+ / -significa que os ratos perdem uma cópia do gene p53 tipo selvagem., Os resultados mostraram que nenhum dos ratinhos com p53+/m desenvolveu tumores potencialmente fatais, no entanto, mais de 80% dos ratinhos com p53+/-e mais de 45% dos ratinhos com p53+/+ desenvolveram estes tipos de tumores. Olhando para o interior dos tumores, foram observadas lesões tumorais localizadas em 2 de 35 ratinhos p53+/m, em contraste, vários tumores como linfomas, e osteossarcomas foram encontrados em ratinhos p53+/- e p53+/+. Durante esta experiência, observaram também que a idade média de p53+/m foi de 96 semanas, enquanto a idade média de p53+/m foi de 116 a 118 semanas., Tyner e colegas de trabalho também examinaram a possibilidade de que a vida mais curta de p53+/m foi associada ao envelhecimento. Eles descobriram que após 18 meses, os ratos p53+/m começaram a perder peso e vigor, como para os ratos p53+ / m, pesos reduzidos foram observados com a idade de 30-36 meses. ratos p53+ / m também exibem lordokifosis. Dependendo da análise de Raio-X, ratinhos p53+/m exibiram densidade óssea reduzida na idade de 12 meses, e se tornará grave com a idade de 18 meses. Este é um símbolo de osteoporose e a osteoporose é um marcador do envelhecimento em seres humanos e ratinhos . Tyner et al., também testou a tolerância ao estresse, uma vez que esta capacidade também é um marcador do envelhecimento . Eles realizaram biópsias de 3 mm de punção na pele das costas de ratos velhos e jovens anestesiados p53+/m e p53+/+. Os resultados mostraram que muitos dos ratinhos p53+/m antigos morreram após a injecção da dose padrão de Avertin, indicando que os ratinhos p53+/m antigos eram menos tolerantes ao stress .em 1991, descobriu-se que p53 tem a capacidade de induzir apoptose, enquanto em 2003, Mihara e outros cientistas descobriram que p53 também tem um papel de apoptose nas mitocôndrias ., Uma vez que algumas proteínas mitocondriais têm a capacidade de activar a apoptose celular, quer por caspases activas, quer neutralizando inibidores citosólicos. No exemplo da caspase induzida pelo citocromo C, após receber o sinal de apoptose, o citocromo c é libertado do espaço intermembranar da mitocôndria, e, por sua vez, liga-se ao Apf-1, que existe como um monómero inactivo, induz a sua alteração conformacional e aumenta a sua afinidade de ligação para o dATP/ATP em 10 vezes do que o Apaf-1 liga o dATP/ATP isoladamente. Então o complexo Apaf-1-citocromo c liga-se a dATP/ATP, formando o apoptossoma., Depois disso, o domínio de recrutamento da caspase (CARD) do Apaf-1 exposto no apoptossoma, recruta a procaspase-9 e depois auto-activa-se. O complexo final então se divide e ativa outras caspases, como a caspase-3, que, por sua vez, clivem moléculas importantes na célula, causando condensação de cromatina, fragmentação de DNA e, finalmente, levando à apoptose . A figura 2 mostra a via de activação da caspase induzida pelo citocromo c.
Mihara M et al. os cientistas descobriram que o gene p53 selvagem pode ser translocado para a superfície mitocondrial das células tumorais rapidamente. No experimento, eles descobriram que algumas proteínas do tipo selvagem p53 induzidas pelo estresse têm a capacidade de translocação para a mitocôndria dos timócitos em células humanas ou de ratinho após apoptose devido a danos no DNA e hipoxia. Em seguida, estas proteínas p53 de tipo selvagem induzem permeabilização das mitocôndrias e causam uma série de alterações que ocorrem nas mitocôndrias, como liberar o citocromo c, formando complexo com Bcl2 e BclXL .,como um bom resultado clínico com pouco efeito secundário, a terapia genética é popular. No final de 2005, existiam 1020 ensaios de terapia genética na base de dados do Journal of Gene Medicine. Entre estes ensaios, 66% das terapias genéticas foram conduzidas em doentes com cancro, e 58 ensaios deste rAd-p53 usado, um adenovírus recombinante que codificou o gene p53 humano. Em abril de 2004, foi lançada formalmente uma injecção de adenovírus humano recombinante (Gendicina). A gendicina é utilizada no tratamento do carcinoma de células escamosas da cabeça e do pescoço e foi aprovada pela State Food and Drug Administration da China em outubro. 16, 2003., Tornou-se o primeiro produto de terapia genética no mundo a ser aprovado pelo governo chinês .o gene p53 foi descoberto para regular o metabolismo em 2005. A fim de transferir da fase G1 para a fase S, as células devem ter suporte suficiente de matérias-primas para o DNA, organelas e síntese de proteínas. Para regular este processo, são necessários alguns postos de controlo. Um deles é o ponto de controlo dependente da glucose em G1/S. é regulado pela proteína cinase activada pela AMP (AMPK). Quando a glicose é esgotada, AMPK pode fosforilar a proteína p53, que por sua vez induz a prisão celular, e evita a morte celular., As células que encontram a paragem dependente do p53 Irão reentrar no ciclo celular quando a glucose for suficiente .
sabe-se que a inactivação de p53 é necessária para a formação de tumores. Bykov et al. VJ e Snydel EL et al. note que o funcionamento inadequado do p53 pode levar à proliferação de um tumor existente . Ventura e seus colegas de trabalho fizeram algumas experiências para testar esta hipótese. Eles restauraram a função de p53 endógena em tumores autóctones primários para examinar a consequência da reativação p53., O resultado mostrou que a reativação p53 foi responsável pela regressão de tumores autóctones. Isso significa que a proteína p53 inactivada pode levar ao desenvolvimento do tumor . Xue e outros cientistas também fizeram uma experiência para testar as consequências da reativação do p53 em tumores. Usaram interferência reversível no ARN (RNAi) para regular a expressão do p53 endógeno em ratinhos com cancro do fígado. No experimento, doxiciclina (Dox) é usada para reativar p53, como a expressão de p53 é totalmente suprimida quando Dox está faltando e rapidamente restaurada quando Dox é adicionado., Quando tratado com Dox, p53 miRNA foi desligado o que, por sua vez, causa aumento da expressão de p53. O resultado mostrou que os tumores em ratinhos tratados com Dox tornam-se indetectáveis após 12 dias, enquanto os tumores em ratinhos não tratados cresceram rapidamente. Para testar a consequência da reactivação transitória de p53, trataram ratos com Dox durante 4 dias e depois pararam. O resultado mostrou que mesmo um tratamento de dois dias pode causar regressão de tumores e 4 dias de tratamento pode causar os tumores a regredir completamente., Eles também apontaram que durante a regressão tumoral, p53 transitoriamente reativado pode desencadear senescência celular, não apoptose. No mesmo ano, a Hu descobriu que a implantação embrionária em ratinhos p53-/- fêmeas é regulada pelo factor inibitório da leucemia (LIF). O LIF é uma citocina secretada e é importante para a implantação do blastocisto. O gene que codifica o LIF é identificado como o gene alvo p53 e o local de ligação p53 está localizado no intron 1 em humanos e ratos .