An automatic procedure written in Microsoft Visual Basic (VB) 6.,0 atualizações de DAVI, semanalmente, com os seguintes procedimentos: a chamada de uma série de Perl e Java, as aplicações que transferir dados públicos através anônimo protocolos de transferência de arquivo (FTP) (Tabela 1); descompactar e analisar anotação desejada de dados; criar delimitado por tabulação de dados, arquivos prontos para importação de base de dados; e importar dados para o Oracle 8i relational database management system (RDBMS), usando o Oracle”s SQL*Loader aplicação. Microsoft”s IIE web server and Active Server Page technology are used to access the databeans using JavaBeans and the structured query language (SQL)., Os números LocusLink para conjuntos de sonda Affymetrix são derivados de associações da Universidade de Michigan ou NetAffx . As anotações funcionais e as referências cruzadas de bases de dados são derivadas da LocusLink, que fornece representações estáveis e curadas de genes. Para informações mais detalhadas sobre as fontes de dados utilizadas por DAVID, consulte a seção FAQ em .,
módulos de Análise
DAVID é composto de quatro módulos principais: Ferramenta de Anotação, GoCharts, KeggCharts, e DomainCharts. A Ferramenta de anotações é um método automatizado para a anotação funcional de listas de genes. Qualquer combinação de dados de anotação pode ser escolhida a partir de 10 opções selecionando as caixas de controle apropriadas (Tabela 2)., As anotações são adicionadas à lista de genes submetidos, selecionando o botão enviar, que retorna uma tabela HTML contendo a lista original do usuário de identificadores anexados com as anotações funcionais escolhidas. Genes não anotados são incluídos na saída sem dados anexados para fins de rastreamento.,
O GoCharts módulo mostra graficamente a distribuição da diferencialmente expressos genes entre as categorias funcionais, usando o vocabulário controlado do Gene Ontology Consortium (GO), que fornece uma linguagem estruturada que pode ser aplicado para as funções de genes e proteínas em todos os organismos, mesmo como o conhecimento continua a acumular-se e mudar ., A linguagem está estruturada em um grafo acíclico direcionado (DAG), onde o termo especificidade aumenta e a cobertura do genoma diminui à medida que se move para baixo a hierarquia. Em contraste com uma verdadeira hierarquia, os Termos filhos em um DAG podem ter mais de um termo pai e podem ter uma classe diferente de relacionamento com seus pais diferentes. A estrutura da GO começa com três categorias principais: processo biológico, função Molecular e componente celular., O processo biológico inclui objetivos biológicos amplos, tais como mitose ou metabolismo de purina, que são realizados por conjuntos ordenados de funções moleculares. A função Molecular descreve as tarefas realizadas por produtos de genes individuais; exemplos são Fator de transcrição e DNA helicase. O tipo de classificação dos componentes celulares envolve estruturas subcelulares, localizações e complexos macromoleculares; exemplos incluem núcleo, telômero e complexo de reconhecimento de origem., Depois de escolher um tipo de classificação, os níveis que determinam a cobertura da lista e a especificidade são escolhidos selecionando o botão de rádio apropriado. O Nível 1 fornece a maior cobertura da lista com a menor quantidade de especificidade de termo. Com cada cobertura de nível crescente diminui enquanto a especificidade aumenta de modo que o nível 5 fornece a menor quantidade de cobertura com a mais alta especificidade de termo.
os dados de classificação são apresentados como um gráfico de barras, onde o comprimento da barra representa o número de identificadores de genes em cada categoria., O usuário pode definir parâmetros de visualização para classificar dados de saída e exibir categorias que contêm pelo menos um número mínimo de genes. Selecionando uma barra individual abre uma nova tabela HTML que mostra o identificador do gene, o número LocusLink, o nome do gene, a classificação atual e outras classificações para cada gene nessa categoria. Um botão ” Mostrar tudo “abre uma nova tabela HTML que mostra todos os dados de classificação e um botão” Mostrar os dados de gráficos ” abre uma tabela HTML que contém os dados de gráfico subjacentes, permitindo assim que os usuários recriem gráficos de gráfico personalizados em um programa de folha de cálculo., Um novo gráfico pode ser exibido para qualquer subconjunto de genes, selecionando o tipo de classificação e nível usando as caixas de controle e botões de rádio disponíveis dentro da página atual do usuário que permitem as capacidades de drill-down. Uma contagem do número de genes anotados é incluída na saída, e genes não anotados são ligados na categoria “não-classificados”, proporcionando assim aos usuários um sistema de rastreamento automatizado para genes não anotados.
KeggCharts apresentam graficamente a distribuição de genes diferentes entre as vias bioquímicas de KEGG., Cada Via está ligada ao mapa da via KEGG, onde genes diferentes da lista original são realçados em vermelho. Nesta visão, os genes estão ainda ligados a anotações adicionais disponíveis através do sistema de recuperação DBGET do KEGG . Tal como acontece com o GoCharts, o usuário pode definir parâmetros de visualização para classificar dados de saída e exibir categorias que contêm pelo menos um número mínimo de genes e a visualização do KeggCharts herda todas as características dinâmicas do GoCharts.
DomainCharts display the distribution of differentially expressed genes among PFAM protein domains ., Cada designação de domínio está ligada à conservada base de dados de domínio (CDD) do National Center for Biotechnology Information (NCBI), onde detalhes sobre a função, estrutura e sequência de domínio estão prontamente disponíveis. Como com GoCharts e KeggCharts, o usuário pode definir parâmetros de visualização para classificação de dados de saída e exibição de categorias que contenham pelo menos um número mínimo de genes e o DomainCharts visualização herda todas as características dinâmicas de GoCharts e KeggCharts. Para mais informações sobre a funcionalidade de DAVID visite a seção FAQ at .,
Using DAVID to mine functional annotation
To demonstrate the functionality of DAVID we analyzed a list of genes differentially expressed in human peripheral blood mononuclear cells (PBMCs) after incubation with HIV-1 envelope proteins. Detalhes dos procedimentos de hibridização experimental, RNA e GeneChip, juntamente com detalhes das normalizações chip-a-chip e análise estatística da expressão diferencial do gene são fornecidos em Cicala et al. ., Resumidamente, as CPSP humanas primárias e os macrófagos derivados de monócitos foram incubados durante 16 horas com a proteína do envelope do VIH-1 (gp120). Microarrays oligonucleotídeos de alta densidade (Affymetrix HU-95A GeneChip) foram utilizados para monitorizar os acontecimentos transcritoriais induzidos pela gp120. Esta análise resultou na identificação de 402 genes diferentes.enquanto 16 genes modulados por gp120 do VIH-1 foram anteriormente associados à replicação do VIH e/ou à sinalização do envelope, os restantes genes têm uma função desconhecida ou nunca foram associados ao VIH-1 ou à gp120., A conversão desta lista de genes em significado biológico requer a recolha de informações pertinentes a partir de vários repositórios de dados. Para muitos pesquisadores, este processo consiste em navegação iterativa através de vários bancos de dados para cada gene, coletando manualmente informações específicas do gene sobre seqüência, função, via e associação de doenças. Em contraste, a abordagem sistemática de DAVID simultaneamente adiciona informações biologicamente ricas derivadas de várias fontes de dados públicas para listas de genes em paralelo., Selecionar a Ferramenta de anotações de DAVID e carregar a lista de 402 genes de expressão diferente inicia a anotação funcional e a análise de todo o conjunto de dados. Uma vez submetida, a lista de genes é armazenada para toda a sessão de análise, permitindo que os usuários mudem entre módulos sem ter que voltar a submeter dados.
Ferramenta de anotação
a Ferramenta de anotação oferece várias opções de anotação e constrói uma vista tabular da lista de genes dos utilizadores e das anotações disponíveis (Tabela 2)., Escolhendo o símbolo genético dos campos de anotação, LocusLink, OMIM, Unigene, sequência de referência e nome do Gene seguido pela seleção do botão “Upload” produz uma tabela HTML no navegador web contendo todos os genes e suas anotações disponíveis, onde identificadores de genes, dados descritivos e de classificação são retirados do banco de dados e anexados à lista de genes (Figura 1). Identificadores de genes como o símbolo genético e a LocusLink estão ligados a dados adicionais específicos de genes disponíveis em suas fontes originais, fornecendo assim detalhes detalhados de genes específicos e pedigrees de anotação., Os dados de classificação e resumos funcionais podem ser usados para pesquisar rapidamente informações relevantes para o sistema experimental do pesquisador. O tempo de servidor necessário para a execução deste módulo correlaciona-se linearmente com o tamanho da lista de genes e leva menos de 45 segundos para listas de até 1.000 genes (Figura 2, números entre parênteses representam valores r2). Estes resultados demonstram a potência e eficiência de uma abordagem integrada à anotação funcional de grandes conjuntos de dados.,

de Saída da Ferramenta de Anotação. Mostrados são anexadas anotações para os primeiros vários conjuntos de sonda Affymetrix em uma tabela HTML contendo todas as 402 entradas. Foram apresentadas informações categóricas sobre as condições experimentais, juntamente com os identificadores do conjunto de sonda Affymetrix e incluídas na saída na coluna de valores. Identificadores como Symbol, LocusLink, OMIM, RefSeq e Unigene accessions são hiper-ligados a suas fontes de origem para informações mais detalhadas., O texto incluído nos campos de resumo é derivado de informações descritivas e funcionais fornecidas nos relatórios LocusLink do NCBI.
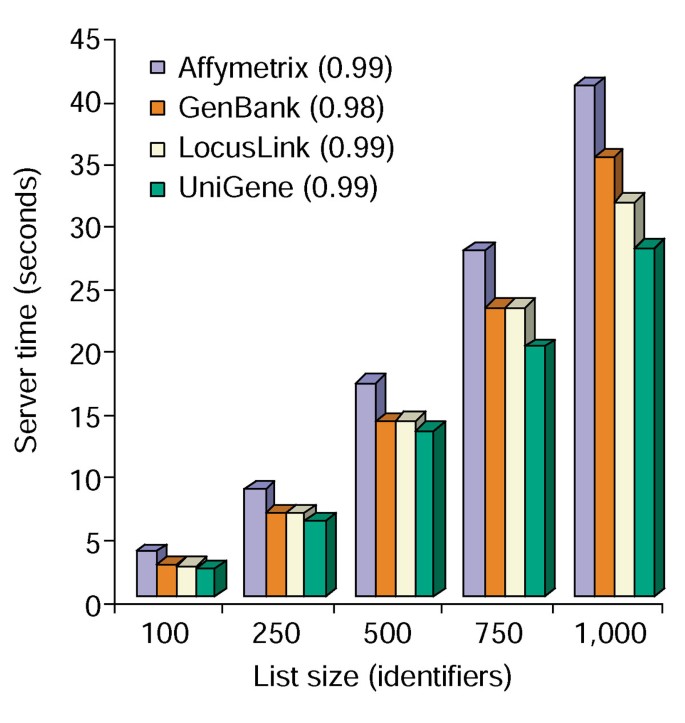
o Tempo de análise da Ferramenta de Anotação. Tempo de servidor necessário (eixo y) para adicionar simultaneamente todas as 10 opções de anotação para listas de genes que variam em tamanho de 100 a 1000 (eixo x)., A média de três ensaios para listas de genes contendo Affymetrix, GenBank, LocusLink e identificadores UniGene são mostrados e os números entre parênteses representam o valor r2 da correlação entre o tamanho da lista de genes e o tempo de servidor necessário para a anotação.
GoCharts
Escolha a GoCharts módulo abre uma nova janela com uma variedade de opções., Os usuários escolhem entre três tipos gerais de classificação (processo biológico, função molecular e componente celular) e cinco níveis de anotação que representam cobertura de termo e especificidade (ver seção módulos de análise). Qualquer combinação de classificação e nível de cobertura pode ser especificado. Também estão incluídas as opções para anotar listas de genes com todos os Termos GO disponíveis ou apenas os termos mais específicos, que são referidos como nós terminais., A opção de escolher diferentes níveis de especificidade de termo proporciona flexibilidade necessária e, assim, permite aos investigadores determinar dinamicamente qual o nível de cobertura e especificidade que melhor se adequa aos seus dados e fase de análise. Por exemplo, as análises em fase inicial podem consistir em anotar listas de genes com termos muito gerais, a fim de obter uma ampla compreensão dos dados. Neste caso, selecionar processo biológico e Nível 1 classifica genes usando termos gerais como” morte “e”comunicação celular”., A utilização de uma maior especificidade a termo facilita a extracção de informações funcionais mais pormenorizadas. Neste caso, selecionando o processo biológico e o nível 5 classifica genes usando termos como” alterações mitocondriais apoptóticas “e”percepção quimiossensorial”.
no entanto, a maior especificidade de termo vem um custo, na medida em que aumenta a cobertura da lista diminui (Figura 3). Em nossos estudos, descobrimos que o Nível 2 tipicamente mantém uma boa cobertura, ao mesmo tempo em que proporciona especificidade de termo significativa., A figura 4a ilustra como a visualização dos GoCharts revela rapidamente que 35 genes de expressão diferente estão envolvidos em”Respostas ao stress”. Cada termo GO pode ser visto na árvore ou vista DAG por hiperlinks para QuickGO .
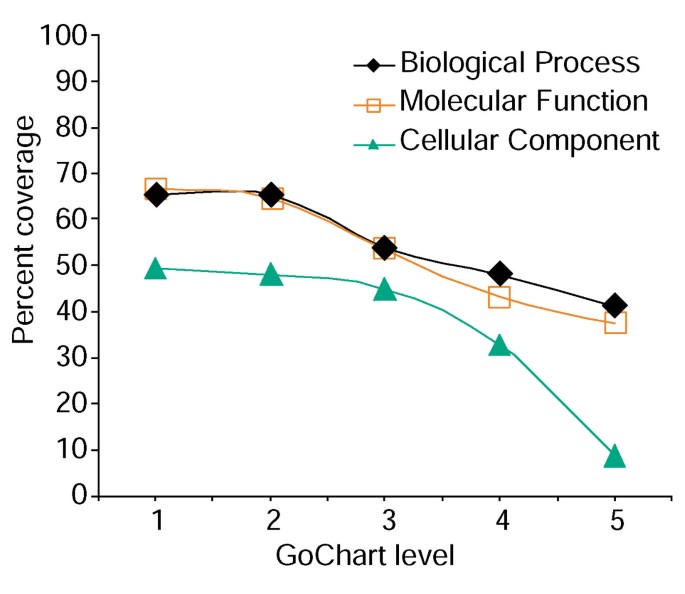
a Análise do gene-lista de cobertura usando GoCharts. A list of 402 Affymetrix probe set identifiers were annotated with the Proteome assigned functional classifications provided by LocusLink., A cobertura percentual representa o número de genes de 402 que foram anotados em um nível de termo-especificidade dentro do processo biológico, função Molecular e tipos de classificação de componentes celulares. A cobertura percentual diminui à medida que a especificidade dos Termos aumenta.
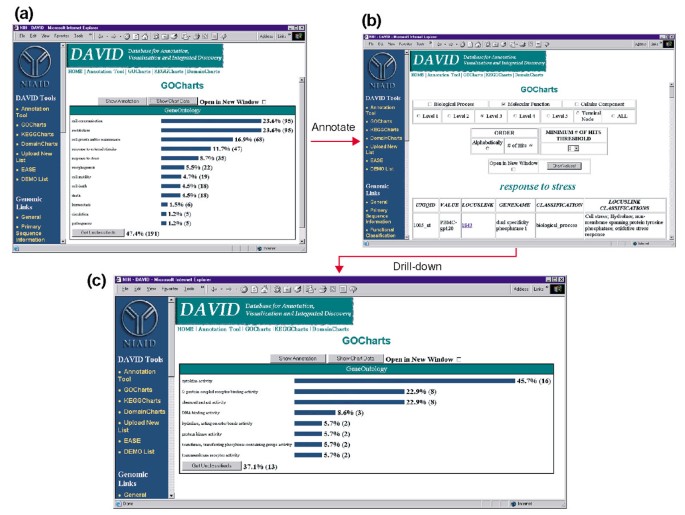
Saída de GoCharts. (a) a bar chart showing the distribution of differentially expressed genes among Gene Ontology (GO) Biological Processes., Os parâmetros foram definidos para ir Nível 2, um limiar de hit de cinco, e a saída foi classificada por hit count. As barras azuis estão ligadas aos dados adicionais de anotação apresentados na alínea b). Selecionando a barra azul em (a) correspondente a “resposta ao estresse” abre uma tabela HTML mostrando o LocusLink, nome do gene, classificação atual e outros dados de classificação para os genes nessa categoria. (C) este subconjunto de genes envolvidos na “resposta ao stress” foi ainda caracterizado pela seleção da função Go Molecular, GO Nível 3, um limiar de hit de 2, e ordenado pela contagem de hit., Selecionando o botão “valores de gráfico” cria um novo histograma revelando que 16 dos 35 genes de resposta a stress codificam proteínas possuindo atividade citocina.,
uma Vez que o HIV-1 tem um grande impacto sobre a função de células do sistema imunológico e sua capacidade de realizar as respostas ao estresse, selecionamos o histograma de barras que representa o número de genes envolvidos na resposta ao estresse, que abre uma tabela HTML que contém o Affymetrix identificador, LocusLink número, nome de gene, a classificação actual, e outras classificações para todos os 35 genes (Figura 4b)., Agora que reduzimos nossa lista de genes para aqueles genes envolvidos em Respostas ao estresse, nós ainda caracterizamos este subconjunto repetindo o procedimento GoCharts disponível no topo da tabela HTML de resposta ao estresse. Escolhendo a função molecular, o Nível 3 produz um novo histograma que rapidamente revela que quase metade (16/35) dos genes de resposta ao stress possuem atividade citocina (figura 4c)., Com efeito, foi demonstrado que as citocinas desempenham um papel importante no ciclo de vida do VIH-1 e os resultados aqui obtidos sugerem que o tratamento de PBMCs com proteínas do envelope do VIH-1 modula significativamente a transcrição de numerosos genes da citocina. A eficiência com que a GoCharts sistematicamente resumiu este grande conjunto de dados com visualizações gráficas, permanecendo ligada a dados primários e recursos externos melhorou drasticamente o processo de descoberta.,
KeggCharts
a figura 5a retrata a produção de KeggCharts com um histograma que mostra a distribuição de genes expressos diferencialmente entre as vias bioquímicas. O gráfico mostra que uma via KEGG de apoptose inclui cinco genes induzidos pela gp120 HIV-1. A selecção do nome da via Abre o correspondente mapa da via bioquímica de KEGG e destaca em traços vermelhos os genes que funcionam de forma diferente nessa via (figura 5b). Nesta visão, os genes estão ainda ligados a anotações adicionais disponíveis através do sistema de recuperação DBGET do KEGG ., Note que apenas quatro genes na Via da apoptose de KEGG são realçados em vermelho, enquanto a ferramenta de KeggCharts mapeou cinco conjuntos de sonda de Affymetrix para a via da apoptose. Esta diferença é devido ao fato de que duas das sondas Affymetrix estão visando o mesmo gene TNF-alfa.
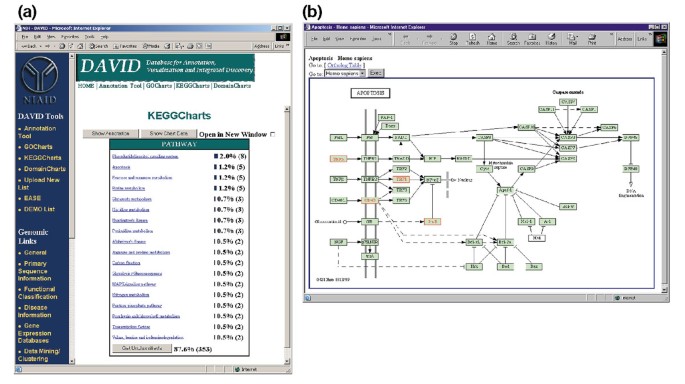
Saída de KeggCharts. (a) Visualization chart showing the distribution of 402 genes among KEGG biochemical pathways. O hit threshold foi definido para três e a saída foi ordenada por hit count., O grande número de identificadores não classificados deve-se ao facto de a KEGG ser centrada nas vias bioquímicas e, por conseguinte, proporcionar uma baixa cobertura das listas de genes. Similarmente à saída de GoCharts, as barras azuis representam o número de genes em cada via. Selecionando uma barra azul abre uma tabela HTML mostrando o LocusLink, nome do gene, classificação atual e outros dados de classificação para os genes nessa via (dados não mostrados)., b) a via bioquímica de KEGG que surge na sequência da Selecção do nome da via “apoptose” na alínea a) retrata quatro genes diferentes expressos na Via da apoptose, destacando-os em verde claro e vermelho. O fato de que a via KEGG destaca apenas quatro genes, enquanto que a KeggChart mapeia cinco conjuntos de sonda Affymetrix para a via apoptose é devido ao fato de que duas sonda define o mesmo gene TNF-alfa.,
DomainCharts
DomainCharts são operacionalmente semelhante para ambos os KeggCharts e GoCharts, exceto que os resultados visualmente retratando a distribuição de genes entre PFAM proteína domínios (Figura 6a). O histograma DomainCharts identifica 16 genes com domínios cinase (pkinase), provavelmente refletindo os efeitos do HIV-1 gp120 na máquina de transdução de sinal. O gráfico também identifica seis genes com domínios de interleucina-8 (IL-8), um domínio que representa um motivo altamente conservado entre as citoquinas de resposta ao estresse., Selecionando o nome de domínio “IL8” abre a página CDD (conserved Domain Database) correspondente a esse domínio PFAM (figura 6b). Esta página fornece uma sequência detalhada, estrutura e informações funcionais sobre o domínio IL-8 e as proteínas que o contêm.
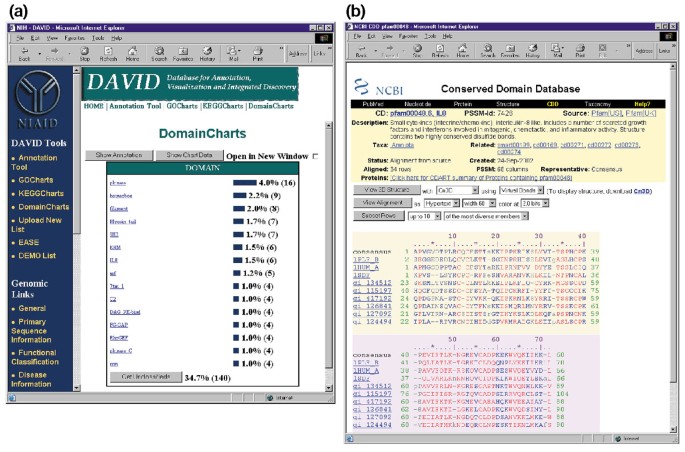
Saída de DomainCharts. (a) Gráfico de visualização que mostra a distribuição de 402 genes entre domínios proteicos. Os parâmetros foram definidos para um limiar mínimo de acerto de quatro e a saída foi ordenada por Contagem de acerto., Semelhante à saída de GoCharts e KeggCharts, as barras azuis representam o número de genes que contêm esse domínio em particular. Selecionando uma barra azul abre uma tabela HTML mostrando o LocusLink, nome do gene, classificação atual e outros dados de classificação para os genes nessa via (dados não mostrados)., (b) Selecionar o nome de domínio “IL8” em (a), que contém seis diferencialmente expressos genes, leva o usuário para uma nova página que contém a saída do Conservada Domínio de Banco de dados (CDD) do NCBI, que fornece informações detalhadas sobre a IL-8 de domínio, incluindo a informação estrutural, vários sequência de alinhamentos e informações descritivas sobre o domínio e as proteínas que possuem.