Eine automatisierte Prozedur geschrieben, die in Microsoft Visual Basic (VB) 6.,0 updates werden wöchentlich mit den folgenden Verfahren durchgeführt: Rufen Sie eine Reihe von Perl-und Java-Anwendungen auf, die öffentliche Daten über anonyme Dateiübertragungsprotokolle (FTP) herunterladen (Tabelle 1); Entpacken und analysieren Sie die gewünschten Annotationsdaten; Erstellen Sie tabulatorgetrennte Datendateien, die für den Datenbankimport bereit sind; und importieren Sie Daten mithilfe der SQL*Loader-Anwendung von Oracle in ein relationales Oracle 8i-Datenbankverwaltungssystem (RDBMS). Der IIE-Webserver und die Active Server-Seitentechnologie von Microsoft werden verwendet, um mit JavaBeans und der Structured Query Language (SQL) auf die Datenbank zuzugreifen., LocusLink-Nummern für Affymetrix-Sondensätze werden von Verbänden der University of Michigan oder NetAffx abgeleitet . Funktionelle Anmerkungen und Datenbank-Querverweise werden von LocusLink abgeleitet, das stabile, vom Menschen kuratierte Darstellungen von Genen bereitstellt. Weitere Informationen zu den von DAVID verwendeten Datenquellen finden Sie im FAQ-Bereich unter .,
Analysemodule
DAVID besteht aus vier Hauptmodulen: Annotation Tool, GoCharts, KeggCharts und DomainCharts. Das Annotation Tool ist eine automatisierte Methode zur funktionellen Annotation von Genlisten. Jede Kombination von Annotationsdaten kann aus 10 Optionen ausgewählt werden, indem die entsprechenden Kontrollkästchen ausgewählt werden (Tabelle 2)., Die Anmerkungen werden der gesendeten Funktionsliste hinzugefügt, indem Sie die Schaltfläche Hochladen auswählen, die eine HTML-Tabelle zurückgibt, die die ursprüngliche Liste der Bezeichner des Benutzers enthält, die an die ausgewählten funktionalen Anmerkungen angehängt sind. Nicht annotierte Gene sind in der Ausgabe ohne angehängte Daten zu Verfolgungszwecken enthalten.,
Das GoCharts-Modul zeigt die Verteilung von differentiell exprimierten Genen unter Funktionskategorien grafisch anhand des kontrollierten Vokabulars des Gene Ontology Consortium (GO) an, das eine strukturierte Sprache bietet, die auf die Funktionen von Genen und Proteinen in allen Organismen angewendet werden kann, auch wenn das Wissen weiter akkumulieren und ändern ., Die Sprache ist in einem gerichteten azyklischen Graphen (DAG) strukturiert, wobei die Termspezifität zunimmt und die Genomabdeckung abnimmt, wenn man sich in der Hierarchie nach unten bewegt. Im Gegensatz zu einer echten Hierarchie können untergeordnete Begriffe in einer DAG mehr als einen übergeordneten Begriff haben und eine andere Klasse von Beziehungen zu ihren verschiedenen Eltern haben. Die Struktur von GO beginnt mit drei Hauptkategorien, biologischem Prozess, molekularer Funktion und zellulärer Komponente., Der biologische Prozess umfasst breite biologische Ziele wie Mitose oder Purinstoffwechsel, die durch geordnete Anordnungen molekularer Funktionen erreicht werden. Molekulare Funktion beschreibt die Aufgaben einzelner Genprodukte; Beispiele sind Transkriptionsfaktor und DNA-Helicase. Der Klassifikationstyp der zellulären Komponente umfasst subzelluläre Strukturen, Standorte und makromolekulare Komplexe; Beispiele hierfür sind Nucleus, Telomere und Origin Recognition Complex., Nach der Auswahl eines Klassifikationstyps werden Ebenen ausgewählt, die die Listenabdeckung und-spezifität bestimmen, indem das entsprechende Optionsfeld ausgewählt wird. Level 1 bietet die höchste Abdeckung Liste mit den geringsten Begriff Spezifität. Mit jedem zunehmenden Level nimmt die Abdeckung ab, während die Spezifität zunimmt, so dass Level 5 die geringste Abdeckung mit der höchsten Termspezifität bietet.
Klassifizierungsdaten werden als Balkendiagramm angezeigt, wobei die Länge des Balkens die Anzahl der Genkennungen in jeder Kategorie darstellt., Der Benutzer kann Visualisierungsparameter zum Sortieren von Ausgabedaten und zum Anzeigen von Kategorien festlegen, die mindestens eine Mindestanzahl von Genen enthalten. Wenn Sie eine einzelne Leiste auswählen, wird eine neue HTML-Tabelle geöffnet, in der die Genkennung, die LocusLink-Nummer, der Genname, die aktuelle Klassifikation und andere Klassifikationen für jedes Gen in dieser Kategorie angezeigt werden. Eine Schaltfläche “ Alle anzeigen „öffnet eine neue HTML-Tabelle mit allen Klassifizierungsdaten und eine Schaltfläche“ Diagrammdaten anzeigen “ öffnet eine HTML-Tabelle mit den zugrunde liegenden Diagrammdaten, sodass Benutzer benutzerdefinierte Diagrammgrafiken in einem Tabellenkalkulationsprogramm neu erstellen können., Ein neues Diagramm kann für jede Teilmenge von Genen angezeigt werden, indem Sie den Klassifizierungstyp und die Ebene mit den Kontrollkästchen und Optionsfeldern auf der aktuellen Seite des Benutzers auswählen, die Drilldown-Funktionen ermöglichen. Eine Zählung der Anzahl der mit Anmerkungen versehenen Gene ist in der Ausgabe enthalten, und nicht annotierte Gene werden in die Kategorie „nicht klassifiziert“ binned, so dass Benutzer mit einem automatisierten Tracking-System für Gene, die nicht mit Anmerkungen versehen.
KeggCharts zeigen grafisch die Verteilung von differentiell exprimierten Genen unter den KEGG biochemischen Bahnen an., Jeder Weg ist mit der KEGG-Wegkarte verknüpft, wobei differentiell exprimierte Gene aus der ursprünglichen Liste rot hervorgehoben sind. In dieser Ansicht sind Gene weiter mit zusätzlichen Anmerkungen verknüpft, die über das DBGET-Abrufsystem von KEGG verfügbar sind . Wie bei GoCharts kann der Benutzer Visualisierungsparameter zum Sortieren von Ausgabedaten und zum Anzeigen von Kategorien festlegen, die mindestens eine minimale Anzahl von Genen enthalten, und die KeggCharts-Visualisierung erbt alle dynamischen Funktionen von GoCharts.
DomainCharts zeigen die Verteilung von differentiell exprimierten Genen unter PFAM-Proteindomänen an ., Jede Domänenbezeichnung ist mit der Konservierten Domänendatenbank (CDD) des National Center for Biotechnology Information (NCBI) verknüpft, in der Details zur Domänenfunktion, – struktur und-reihenfolge leicht verfügbar sind. Wie bei GoCharts und KeggCharts kann der Benutzer Visualisierungsparameter zum Sortieren von Ausgabedaten und zum Anzeigen von Kategorien festlegen, die mindestens eine minimale Anzahl von Genen enthalten, und die DomainCharts-Visualisierung erbt alle dynamischen Funktionen von GoCharts und KeggCharts. Weitere Informationen zur Funktionalität von DAVID finden Sie im FAQ-Bereich unter .,
Unter Verwendung von DAVID to mine Functional Annotation
Um die Funktionalität von DAVID zu demonstrieren, analysierten wir eine Liste von Genen, die nach Inkubation mit HIV-1-Hüllproteinen differentiell in menschlichen peripheren mononukleären Blutzellen (PBMCs) exprimiert wurden. Details der experimentellen, RNA-Präparation, und GeneChip-Hybridisierungsverfahren, zusammen mit Details der Chip-zu-Chip-Normalisierungen und statistische Analyse der differentiellen Genexpression sind in Cicala et al. ., Kurz gesagt, primäre humane PBMCs und Monozyten-abgeleitete Makrophagen wurden 16 Stunden lang mit HIV-1-Hüllprotein (gp120) inkubiert. Hochdichte Oligonukleotid-Mikroarrays (Affymetrix HU-95A GeneChip) wurden zur Überwachung gp120-induzierter Transkriptionsereignisse verwendet. Diese Analyse führte zur Identifizierung von 402 differentiell exprimierten Genen.
Während 16 Gene, die durch HIV-1 gp120 moduliert wurden, zuvor mit der HIV-Replikation und/oder Hüllsignalisierung assoziiert waren, sind die verbleibenden Gene von unbekannter Funktion oder wurden nie mit HIV-1 oder gp120 assoziiert., Die Umwandlung dieser Liste von Genen in biologische Bedeutungen erfordert das Sammeln relevanter Informationen aus mehreren Datenrepositorien. Für viele Forscher besteht dieser Prozess aus dem iterativen Durchsuchen mehrerer Datenbanken für jedes Gen und dem manuellen Sammeln von genspezifischen Informationen zu Sequenz, Funktion, Weg und Krankheitsassoziation. Im Gegensatz dazu fügt der systematische Ansatz von DAVID gleichzeitig biologisch reichhaltige Informationen, die aus mehreren öffentlichen Datenquellen stammen, Listen von Genen parallel hinzu., Die Auswahl DAVID Annotation Tool und das Hochladen der Liste der 402 differentiell exprimierten Gene initiiert die funktionelle Annotation und Analyse des gesamten Datensatzes. Nach dem Senden wird die Genliste für die gesamte Analysesitzung gespeichert, sodass Benutzer zwischen Modulen wechseln können, ohne Daten erneut vorlegen zu müssen.
Annotation Tool
Das Annotation Tool bietet mehrere Annotationsoptionen und erstellt eine tabellarische Ansicht der Benutzerliste und der verfügbaren Annotationen (Tabelle 2)., Wenn Sie die Annotationsfelder Gensymbol, LocusLink, OMIM, Unigene, Referenzsequenz und Genname auswählen, gefolgt von der Schaltfläche „Hochladen“, wird im Webbrowser eine HTML-Tabelle mit allen Genen und ihren verfügbaren Annotationen erstellt, in der Genkennungen, Beschreibungs-und Klassifizierungsdaten aus der Datenbank abgerufen und an die Genliste angehängt werden (Abbildung 1). Genkennungen wie Gensymbol und LocusLink sind mit zusätzlichen genspezifischen Daten verknüpft, die an ihren ursprünglichen Quellen verfügbar sind, wodurch detaillierte genspezifische Details und Annotations-Stammbäume bereitgestellt werden., Klassifizierungsdaten und Funktionszusammenfassungen können verwendet werden, um schnell nach Informationen zu suchen, die für das experimentelle System des Forschers relevant sind. Die Serverzeit, die für die Ausführung dieses Moduls benötigt wird, korreliert linear mit der Größe der Genliste und dauert weniger als 45 Sekunden für Listen mit bis zu 1.000 Genen (Abbildung 2, Zahlen in Klammern stellen r2-Werte dar). Diese Ergebnisse zeigen die Leistungsfähigkeit und Effizienz eines integrierten Ansatzes für die funktionale Annotation großer Datensätze.,

Ausgang des Annotationswerkzeugs. Gezeigt werden angehängte Anmerkungen für die ersten Affymetrix Probe-Sets in einer HTML-Tabelle, die alle 402 Einträge enthält. Kategorische Informationen zu den Versuchsbedingungen wurden zusammen mit den Affymetrix-Sondensatzkennungen übermittelt und in die Ausgabe in der Spalte Wert aufgenommen. Kennungen wie Symbol -, LocusLink -, OMIM -, RefSeq-und Unigene-Zugriffe sind für detailliertere Informationen mit ihren Ursprungsquellen verknüpft., Der in den Zusammenfassungsfeldern enthaltene Text wird aus beschreibenden, funktionalen Informationen abgeleitet, die in den LocusLink-Berichten von NCBI bereitgestellt werden.
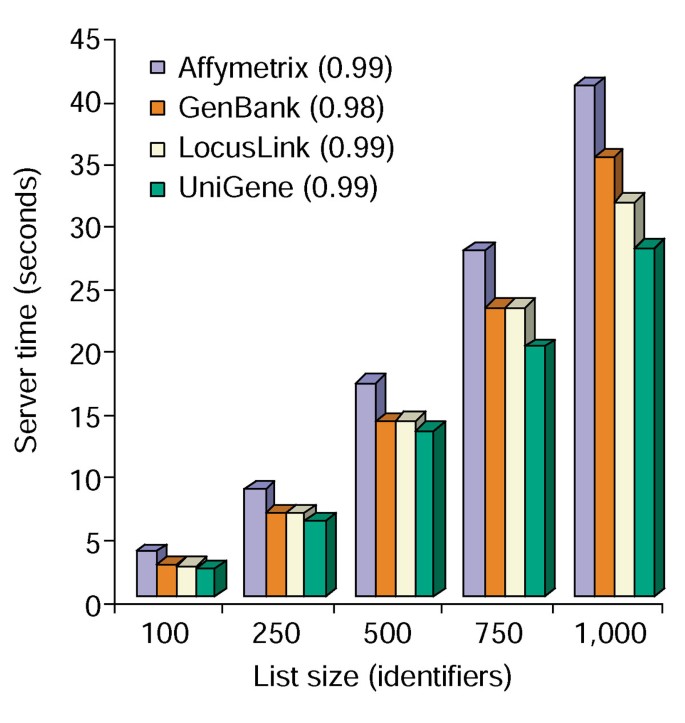
Time analysis of Annotation Tool. Server benötigte Zeit (y-Achse) gleichzeitig append alle 10 Beschriftungs-Optionen, um gen-Listen-Sie reichen in der Größe von 100 zu 1000 (x-Achse)., Der Durchschnitt von drei Versuchen für Genlisten, die Affymetrix -, GenBank -, LocusLink-und UniGene-Kennungen enthalten, wird angezeigt, und die Zahlen in Klammern stellen den Wert der Korrelation zwischen der Größe der Genliste und der für die Annotation erforderlichen Serverzeit dar.
GoCharts
Die Auswahl des GoCharts-Moduls öffnet ein neues Fenster mit einer Vielzahl von Optionen., Benutzer wählen zwischen drei allgemeinen Klassifizierungsarten (biologischer Prozess, molekulare Funktion und zelluläre Komponente) und fünf Annotationsebenen, die Termabdeckung und Spezifität darstellen (siehe Abschnitt Analysemodule). Jede Kombination von Klassifikation und Deckungsgrad kann angegeben werden. Ebenfalls enthalten sind Optionen zum Kommentieren von Genlisten mit allen verfügbaren GO-Begriffen oder nur den spezifischsten Begriffen, die als Terminalknoten bezeichnet werden., Die Möglichkeit, verschiedene Ebenen der Termspezifität auszuwählen, bietet die erforderliche Flexibilität und ermöglicht es den Forschern, dynamisch zu bestimmen, welcher Erfassungsgrad und welche Spezifität am besten zu ihren Daten und dem Analysestudium passen. So können frühe Analysen beispielsweise darin bestehen, Genlisten mit sehr allgemeinen Begriffen zu kommentieren, um ein breites Verständnis der Daten zu erlangen. In diesem Fall klassifiziert der biologische Prozess und die Ebene 1 Gene unter Verwendung allgemeiner Begriffe wie“ Tod „und“Zellkommunikation“., Die Verwendung einer erhöhten Termspezifität erleichtert die Extraktion detaillierterer Funktionsinformationen. In diesem Fall klassifiziert der biologische Prozess und Level 5 Gene unter Verwendung von Begriffen wie“ apoptotische mitochondriale Veränderungen „und“chemosensorische Wahrnehmung“.
Eine erhöhte Termspezifität verursacht jedoch Kosten, da sie mit zunehmender Listenabdeckung abnimmt (Abbildung 3). In unseren Studien stellen wir fest, dass Level 2 typischerweise eine gute Abdeckung beibehält und gleichzeitig eine aussagekräftige Begriffsspezifität bietet., Abbildung 4a zeigt, wie die GoCharts-Visualisierung schnell zeigt, dass 35 differentiell exprimierte Gene an „Stressreaktionen“beteiligt sind. Jeder GO-Begriff kann in den Baum-oder DAG-Ansichten durch Hyperlinks zu QuickGO angezeigt werden .
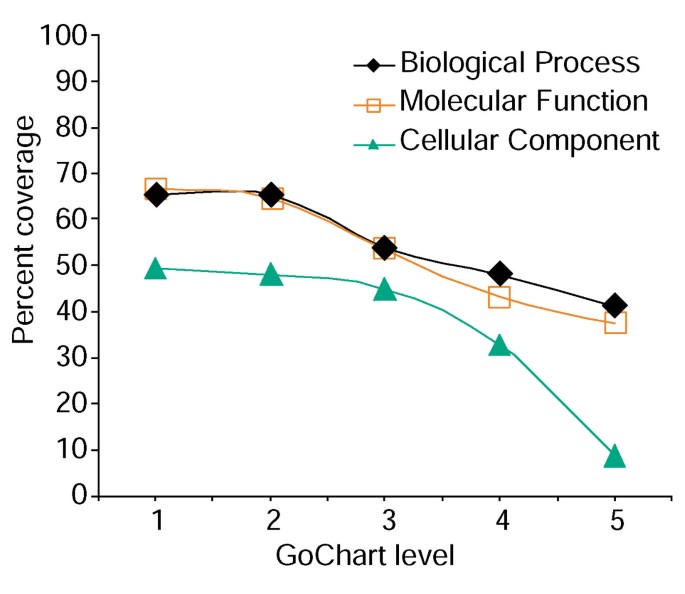
Analyse der Genlistenabdeckung mit GoCharts. Eine Liste von 402 Affymetrix-Sondensatzkennungen wurde mit den von LocusLink bereitgestellten Proteom zugeordneten Funktionsklassifizierungen kommentiert., Prozentabdeckung repräsentiert die Anzahl der Gene von 402, die auf einer Termspezifitätsebene innerhalb der Klassifikationstypen Biologischer Prozesse, molekularer Funktionen und zellulärer Komponenten annotiert wurden. Die Prozentabdeckung nimmt ab, wenn die Termspezifität zunimmt.
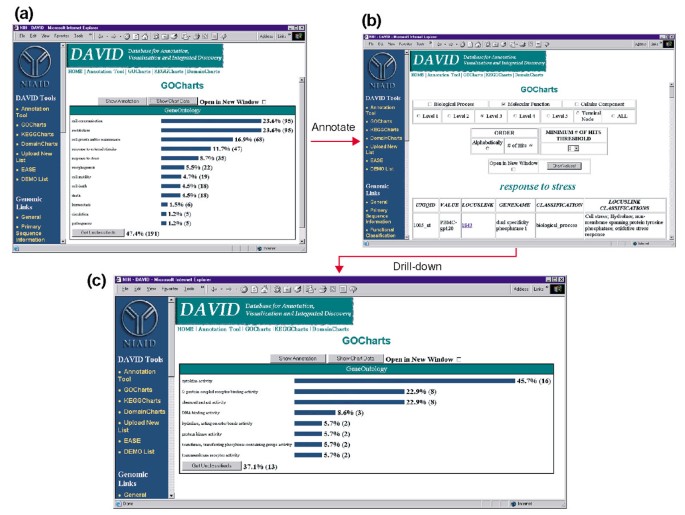
Ausgang von GoCharts. (a) Ein Balkendiagramm, das die Verteilung von differentiell exprimierten Genen auf biologische Prozesse der Gen Ontologie (GO) zeigt., Die Parameter wurden auf Stufe 2 gesetzt, eine Trefferschwelle von fünf, und die Ausgabe wurde nach Trefferanzahl sortiert. Blaue Balken sind mit zusätzlichen Annotationsdaten verknüpft, die in (b) angezeigt werden. Wenn Sie den blauen Balken in (a) auswählen, der „Reaktion auf Stress“ entspricht, wird eine HTML-Tabelle geöffnet, die den LocusLink, den Gennamen, die aktuelle Klassifizierung und andere Klassifizierungsdaten für die Gene in dieser Kategorie anzeigt. (c) Diese Teilmenge von Genen, die an der „Stressreaktion“ beteiligt sind, wurde weiter charakterisiert, indem GO Molecular Function, GO Level 3, eine Trefferschwelle von 2 ausgewählt und nach Trefferzahl sortiert wurden., Wenn Sie die Schaltfläche „Diagrammwerte“ auswählen, wird ein neues Histogramm erstellt, aus dem hervorgeht, dass 16 der 35 Stressreaktionsgene Proteine kodieren, die Zytokinaktivität besitzen.,
Da HIV-1 einen großen Einfluss auf die Funktion von Zellen des Immunsystems und ihre Fähigkeit zur Durchführung von Stressreaktionen hat, haben wir den Histogrammbalken ausgewählt, der die Anzahl der an der Stressreaktion beteiligten Gene darstellt und eine HTML-Tabelle öffnet, die die Affymetrix-Kennung, die LocusLink-Nummer, den Gennamen, die aktuelle Klassifikation und andere Klassifikationen für alle 35 Gene enthält (Abbildung 4b)., Nachdem wir nun unsere Genliste auf jene Gene reduziert haben, die an Stressreaktionen beteiligt sind, haben wir diese Teilmenge weiter charakterisiert, indem wir das oben in der HTML-Tabelle für Stressreaktionen verfügbare GoCharts-Verfahren wiederholt haben. Bei der Wahl der molekularen Funktion erzeugt Level 3 ein neues Histogramm, das schnell zeigt, dass fast die Hälfte (16/35) der Stressreaktionsgene eine Zytokinaktivität besitzt (Abbildung 4c)., Es wurde gezeigt, dass Zytokine eine wichtige Rolle im HIV-1-Lebenszyklus spielen, und die hier erzielten Ergebnisse legen nahe, dass die Behandlung von PBMC mit HIV-1-Hüllproteinen die Transkription zahlreicher Zytokingene signifikant moduliert. Die Effizienz, mit der GoCharts diesen großen Datensatz systematisch mit grafischen Visualisierungen zusammenfasste und gleichzeitig mit Primärdaten und externen Ressourcen verknüpft blieb, verbesserte den Erkennungsprozess drastisch.,
KeggCharts
Abbildung 5a zeigt die Ausgabe von KeggCharts mit einem Histogramm, das die Verteilung differentiell exprimierter Gene auf biochemische Bahnen anzeigt. Die Grafik zeigt, dass ein KEGG-Weg der Apoptose fünf Gene umfasst, die durch HIV-1 gp120 induziert werden. Die Auswahl des Wegnamens öffnet die entsprechende KEGG Biochemical Pathway Map und hebt in rotem Umriss die differentiell exprimierten Gene hervor, die in diesem Weg funktionieren (Abbildung 5b). In dieser Ansicht sind Gene weiter mit zusätzlichen Anmerkungen verknüpft, die über das DBGET-Abrufsystem von KEGG verfügbar sind ., Beachten Sie, dass nur vier Gene im KEGG-Apoptose-Pfad rot hervorgehoben sind, während das KeggCharts-Tool fünf Affymetrix-Sondensätze dem Apoptose-Pfad zugeordnet hat. Dieser Unterschied ist darauf zurückzuführen, dass zwei der Affymetrix-Probesets auf dasselbe „TNF-alpha“ – Gen abzielen.
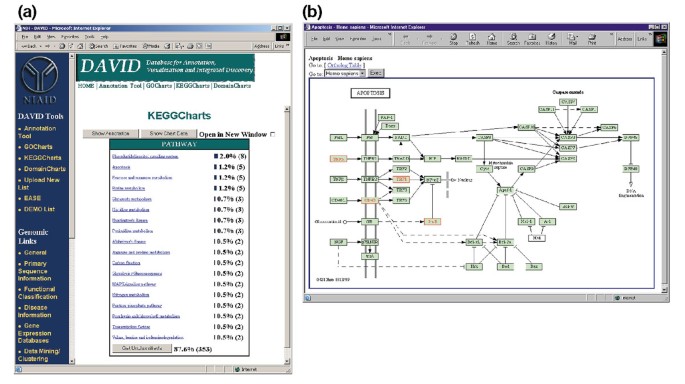
Ausgang von KeggCharts. (a) Visualisierungstabelle, die die Verteilung von 402 Genen unter den biochemischen KEGG-Bahnen zeigt. Die Trefferschwelle wurde auf drei gesetzt und die Ausgabe wurde nach Trefferanzahl sortiert., Die große Anzahl nicht klassifizierter Identifikatoren ist darauf zurückzuführen, dass KEGG biochemisch-pathway-zentriert ist und somit eine geringe Abdeckung von Genlisten bietet. Ähnlich wie bei der Ausgabe von GoCharts stellen blaue Balken die Anzahl der Gene in jedem Pfad dar. Wenn Sie einen blauen Balken auswählen, wird eine HTML-Tabelle geöffnet, die den LocusLink, den Gennamen, die aktuelle Klassifizierung und andere Klassifizierungsdaten für die Gene in diesem Pfad anzeigt (Daten werden nicht angezeigt)., (b) Der biochemische KEGG-Weg, der nach der Auswahl des Wegnamens „Apoptose“ in (a) erscheint, zeigt vier differentiell exprimierte Gene innerhalb des Apoptose-Weges, indem er sie in hellgrün und rot hervorhebt. Die Tatsache, dass der KEGG-Pfad nur vier Gene hervorhebt, während der KeggChart fünf Affymetrix-Sondensätze dem Apoptose-Pfad zuordnet, ist darauf zurückzuführen, dass zwei Sondensätze auf dasselbe „TNF-alpha“ – Gen abzielen.,
DomainCharts
DomainCharts ähneln operativ sowohl KeggCharts als auch GoCharts, mit der Ausnahme, dass die Ergebnisse die Verteilung von Genen unter PFAM-Proteindomänen visuell darstellen (Abbildung 6a). Das DomainCharts-Histogramm identifiziert 16 Gene mit Kinase-Domänen (Pkinase), die wahrscheinlich die Auswirkungen von HIV-1 gp120 auf die Signaltransduktionsmaschinerie widerspiegeln. Das Diagramm identifiziert auch sechs Gene mit Interleukin-8-Domänen (IL-8), einer Domäne, die ein stark konserviertes Motiv unter Stressreaktionszytokinen darstellt., Wenn Sie den Domänennamen „IL8“ auswählen, wird die CDD-Seite (Conserved Domain Database) geöffnet, die dieser PFAM-Domäne entspricht (Abbildung 6b). Diese Seite enthält detaillierte Sequenz -, Struktur-und Funktionsinformationen über die IL-8-Domäne und die darin enthaltenen Proteine.
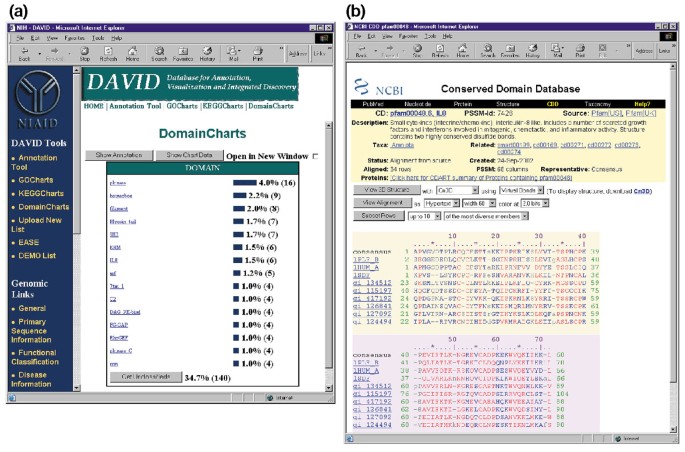
Ausgang von DomainCharts. (a) Visualisierungstabelle, die die Verteilung von 402 Genen unter Proteindomänen zeigt. Die Parameter wurden auf eine minimale Trefferschwelle von vier eingestellt und die Ausgabe wurde nach Trefferanzahl sortiert., Ähnlich wie bei der Ausgabe von GoCharts und KeggCharts stellen blaue Balken die Anzahl der Gene dar, die diese bestimmte Domäne enthalten. Wenn Sie einen blauen Balken auswählen, wird eine HTML-Tabelle geöffnet, die den LocusLink, den Gennamen, die aktuelle Klassifizierung und andere Klassifizierungsdaten für die Gene in diesem Pfad anzeigt (Daten werden nicht angezeigt)., (b) Die Auswahl des Domänennamens „IL8“ in (a), der sechs differentiell exprimierte Gene enthält, bringt den Benutzer auf eine neue Seite, die die Ausgabe aus der konservierten Domänendatenbank (CDD) von NCBI enthält, die detaillierte Informationen über die IL-8-Domäne enthält, einschließlich struktureller Informationen, mehrerer Sequenzausrichtungen und beschreibender Informationen über die Domäne und die Proteine, die sie besitzen.