Einführung
Die Ferkelsterblichkeit ist eines der Hauptselektionsmerkmale in der Schweinezucht und wird von der Sau, den Ferkeln und der Umwelt beeinflusst. Daher ist die Ferkelsterblichkeit ein komplexer Phänotyp und hängt von der Fähigkeit der Sau ab, ihre Nachkommen aufzuziehen, ist aber auch eine Funktion des Geburtsgewichts, des Managements und der Selektion (Knol et al., 2002)., Allerdings tragen auch monogene rezessive Defekte zur Ferkelsterblichkeit bei, obwohl in der Vergangenheit nur wenige Beispiele berichtet wurden (Murgiano et al., 2012; Matika et al., 2019). Selbst in den Fällen, in denen die Wirkung der Mutation stark ist, wird die effiziente Auswahl gegen eine solche Mutation durch die niedrige Frequenz behindert. Bei vielen schweren Defekten sterben Zygoten sehr früh in der Schwangerschaft und hinterlassen keine Spur außer dem Fehlen von Homozygoten in der Population (Derks et al., 2019).,
Inzuchteffekte in kommerziellen Schweinepopulationen werden in der Regel durch selektive Zucht auf verminderte Mortalität bei Ferkeln durch Verbesserung sowohl der Mottenfähigkeit als auch der Lebensfähigkeit von Ferkeln überprüft (Olijslagers, 2018). Varianten, die rezessiven monogenen Defekten zugrunde liegen, werden jedoch innerhalb der Zuchtwerte nicht gut erfasst und driften möglicherweise aufgrund intensiver Selektion zu höheren Frequenzen (Georges et al., 2019). Darüber hinaus können diese Varianten auch als Ergebnis einer Ausgleichsauswahl für einen korrelierten positiven Effekt im heterozygoten Zustand beibehalten werden (Derks et al., 2018).,
Rezessive Defekte tragen nur marginal zur allgemeinen Ferkelsterblichkeit bei (Alonso-Spilsbury et al., 2007). Dennoch sind Varianten, die die Ferkelsterblichkeit beeinflussen, von großer Bedeutung, da diese Varianten die Produktion und den Tierschutz direkt beeinflussen (Baxter et al., 2013; Rutherford et al., 2013). Im Tierpopulationsmanagement ist das niederfrequente Auftreten von Defekten jedoch in der Regel schlecht dokumentiert (oft werden sehr allgemeine Begriffe verwendet), und Syndrome werden oft erst erkannt, wenn sie eine hohe Häufigkeit erreicht haben., Dies ist besonders relevant für Syndrome, die nicht zu sehr unterschiedlichen Phänotypen führen. Daher kann auch in kommerziellen Zuchtpopulationen nur wenig nachverfolgt werden, was spezifische Syndrome betrifft, und die effektive Auswahl gegen spezifische niederfrequente Syndrome erfordert daher neue Ansätze.
In dieser Arbeit beschreiben wir die Entdeckung eines stark schwächenden Syndroms in einer kommerziellen Schweinepopulation durch eine Umfrage, die auf kombinierten SNP-Arrays mittlerer Dichte und Ganzgenomsequenzierung (WGS) basiert., Die Umfrage führte zur Identifizierung einer 16-bp-Frameshift-Deletion im SPTBN4-Gen mit vorhergesagten klaren phänotypischen Folgen bei Homozygoten. Die Trägerfrequenz beträgt in der untersuchten Population etwa 9% und betrifft etwa 0,81% der Populationswürfe. Die Häufigkeit war ausreichend niedrig, um unbekannt zu sein, um eine genetische Grundlage zu haben, und sogar effektiv als spezifisches Syndrom überhaupt nicht erkannt zu werden. Bei der Durchführung der Umfrage wurde eine schwangere Sau von einem Trägerschwein gezeugt., Die betroffenen Ferkel leiden an Myopathie und können nicht laufen, was in der Regel innerhalb weniger Stunden nach der Geburt zum Tod führt, ganz im Einklang mit der vorhergesagten Pathologie im Vergleich zu ähnlichen Fällen bei Menschen und Mäusen.
Material und Methoden
Tiere, Genotypen und Vorverarbeitung
Der Datensatz besteht aus 31.839 Tieren aus einer synthetischen Eberlinie mit großem weißen Hintergrund. Die Linie wird in Topigs Norsvin Nucleus Farms gepflegt und gezüchtet, hauptsächlich basierend auf Produktions-und Gesundheitsmerkmalen., Die Tiere wurden mittels der Illumina GeneSeek benutzerdefinierte 50K SNP chip (Lincoln, NE, USA). Tiere mit einer Häufigkeit fehlender Genotypen > 0.15 wurden entfernt. Wir haben Marker verworfen, die folgende Filterkriterien nicht erfüllten: Eine minimale Anrufrate von 0,85, eine geringe Allelfrequenz > 0,01 und ein Hardy-Weinberg-Test p-Wert unter P < 10-12. Darüber hinaus wurden Marker mit unbekannter Position auf dem Sscrofa11.1-Genomaufbau verworfen, wobei 41.573 Marker nach dem Filtern zurückblieben., Alle Schritte wurden in Plink v1.90b3.30 durchgeführt (Purcell et al., 2007).
Haplotyp-Phasing und Identifizierung des SSC6-Haplotyps
Wir führten Haplotyp-Phasing und Imputation fehlender Stellen in Beagle5.0 durch, wobei der Parameter für die effektive Populationsgröße auf 100 eingestellt war, andere Einstellungen waren Standard (Browning et al., 2018). Erwartete Homozygoten wurden basierend auf der Haplotypfrequenz nach dem Hardy-Weinberg-Prinzip geschätzt. Ein exakter Binomialtest wurde angewendet, um die Anzahl der beobachteten Homozygoten mit der Anzahl der erwarteten Homozygoten zu testen., Der Haplotyp wurde als signifikant erschöpft angesehen, wenn P < 5 × 10-3.
Phänotypische Effekte im Zusammenhang mit dem SSC6-Haplotyp
Wir untersuchten den SSC6-Haplotyp auf Aufzeichnungen über Gesamtzahl der Geborenen, Anzahl der Totgeborenen, mumifizierten Ferkel, Abferkelüberleben und Laktationsüberleben (Überleben bis zu etwa 21 Tagen) von insgesamt 9.666 Würfen. Wir haben diese Phänotypen für alle identifizierten CxC-und CxN-Würfe aufgelistet. Wir haben einen Welch-T-Test verwendet, um zu beurteilen, ob sich die Phänotypen der CxC-Würfe signifikant von den CxN-Würfen unterscheiden. Ein p-Wert < 0.,05 wurde als signifikant angesehen.
Vollgenomsequenzierungsanalyse und Kandidatenvariantenidentifikation
Der Datensatz besteht aus 71 vollgenomsequenzierten Individuen aus der untersuchten Population. Alle 71 Proben waren auch in unserem Datensatz von 31.839 Tieren vorhanden, die auf dem 50K genotypisiert waren. Die 71 Proben haben ein Gesamtvolumen von 1.93 Tbp (Tera-Basenpaare) von 14.16 Milliarden 150-bp-gepaarten Endlesungen (Tabelle S3). Die Proben wurden auf Illumina HiSeq 2000 sequenziert. Wir haben die Sequenzen mit BWA-MEM Version 0.7 auf den Sscrofa11.1-Genom-Build ausgerichtet.,15 (Li und Durbin, 2009) mit einer durchschnittlichen Abbildbarkeit von 98,9% und einer Stichprobenabdeckung von 8,8 bis 14,8 X (10,9 X Durchschnitt). Samblaster wurde verwendet, um PCR-Duplikate zu entfernen (Faust und Hall, 2014). Samtools wurde zum Sortieren, Zusammenführen und Indizieren von bam-Dateien verwendet (Li et al., 2009). Mapping – und Qualitätsstatistiken wurden mit Qualimap generiert (Okonechnikov et al., 2016). Variant Aufruf wurde mit Freebayes v1.1.0 mit folgenden Einstellungen durchgeführt:-min-Basis –Qualität 10-min-Alternative –Fraktion 0.2-haplotype –Länge 0-min-Alternative-count 2 (Garrison und Marth, 2012)., Varianten mit Phred quality score < 20 verworfen wurden (Li et al., 2009). Varianten wurden mit dem Ensemble variant effect predictor (VEP, Release 96) kommentiert (Mclaren et al., 2016). Der Einfluss von Missense-Varianten wurde unter Verwendung von Sortierintoleranz von Tolerant (SIFT) vorhergesagt (Kumar et al., 2009). Die LD-Analyse wurde mit Plink v1.90b3.30 (Purcell et al., 2007) mit folgenden Einstellungen –chr-set 18, –r2 ld-Fenster-r2 0.8.
SPTBN4 Protein-Ausrichtung
Protein-Ausrichtung zwischen der Wildtyp-und mutierten proteins erfolgte mit ClustalO (Madeira et al.,, 2019) und visualisiert mit ESPript 3 (Robert und Gouet, 2014). Weitere Visualisierung und die Validierung erfolgte mit dem JBrowse Genom-viewer-version 1.12.1 (Skinner et al., 2009).
Die Validierung der ursächlichen 16 bp SPTBN4-Deletion
PCR erfolgte unter Verwendung von 60 ng genomischer DNA mit 0,4 µm je Primer, 1,8 mM MgCl2 und 25 Einheiten/ml OneTaq® DNA-Polymerase (OneTaq® 2X Master Mix mit Standardpuffer, New England Biolabs) im PCR-Puffer des Herstellers in einem Endvolumen von 12 µl., Auf die anfängliche Denaturierung für 1 min bei 95°C folgten 35 Zyklen von 95°C für 30 s, 55°C für 45 s, 72°C 90 s, gefolgt von einer 5-min-Verlängerung von 72°C. PCR-Primer für SPTBN4 sind TCAAGGGTGCAGGCTCTTTC vorwärts und GGTAGGAAGCTCGAAGTGGG rückwärts. Der Forward-Primer wurde entweder mit 6-FAM farbstoffmarkiert, um ein fluoreszenzmarkiertes PCR-Produkt zu erzeugen, das auf dem ABI 3730 DNA-Sequenzer nachweisbar ist (Applied Biosystems). Fragmentgrößen wurden mit der GeneMapper Software 5 von ABI bestimmt.,
Histopathologische Untersuchung
Zwei betroffene Ferkel weniger als 1 Woche alt wurden zur Untersuchung an die Pathology Department of Royal Animal Health (Deventer) geschickt. Makroskopisch befanden sich alle Beobachtungen innerhalb normaler Grenzen. Skelettmuskel des Vorderbeins, des Rückenmuskels und des hinteren Beins beider Tiere wurde für routinemäßige H&E Färbung und PTAH Färbung abgetastet. Das Muskelgewebe wurde in getrennten Gläsern gelagert und in Formaldehydlösung 4%, gepuffert (=Formalinlösung 10%, gepuffert) fixiert., Danach wurde das Gewebe in Paraffin eingebettet und gemäß dem Standardoperationsverfahren (SOP RAH) in 2 µm geschnitten. Danach wurden die Folien deparaffinisiert und routinemäßig für Hämatoxylin und Eosin (H&E) in einer automatischen Farbmaschine gefärbt. Gleichzeitig wurden zusätzliche Dias von 2 µm des Muskelgewebes sowie ein positiver Kontrolldias des Muskelgewebes für die manuelle Färbung mit „Phosphotungstinsäure Hämatoxylin“, abgekürzt PTAH, vorbereitet. Diese Färbung wird bevorzugt, um Querschnitte der Skelettmuskulatur zu demonstrieren.,
Zuchtwerte und Assoziationsanalyse
In dieser Studie haben wir 63 im Zuchtprogramm verwendete Merkmale ausgewertet. Deregressed geschätzte zuchtwerte (DEBV) wurden als abhängige Variablen für jedes Merkmal wird im Rahmen der Studie. Der geschätzte Zuchtwert (Estimated Breeding Value, EBV) aller bewerteten Merkmale wurde unter Verwendung der von Garrick et al. (2009). Die EBV jedes Tieres wurde aus der routinemäßigen genetischen Bewertung durch ein kommerzielles Zuchtprogramm (Topigs Norsvin) unter Verwendung eines Tiermodells erhalten., Die reliabilitäten pro Tier für die Zwecke der deregression extrahiert wurden von der genetischen Bewertung basiert auf der Methodik des Tier-und Meyer (2004). Die für die Deregression verwendeten Erbschaften wurden ebenfalls aus der routinemäßigen genetischen Auswertung extrahiert. Schließlich wurden auch Gewichtungsfaktoren auf der Grundlage der geschätzten Zuverlässigkeit des DEBV nach Garrick et al. (2009) mit einem Wert von 0,5 für den Skalar c. Um die Qualität der DEBV zu gewährleisten, nur Tiere mit einem Gewichtungsfaktoren größer als Null und eine Zuverlässigkeit der DEBV größer als 0. ,20 wurden in der Vereinigung analysiert. Die Zuverlässigkeit des DEBV wurde ebenfalls nach Garrick et al. (2009).
Assoziationsanalysen wurden mit der Software ASREML durchgeführt (Gilmour et al.,, 2009) Anwendung des folgenden linearen gemischten Tiermodells:
wobei DEBVij das beobachtete DEBV für das Tier j ist, w der Gewichtungsfaktor für den Rest ist, µ das allgemeine DEBV-Mittel der Population ist, Ri der Trägerstatus (Anzahl des schädlichen Allels) der 4 Mutation i ist, aj der additive genetische Effekt ist, der unter Verwendung einer stammbaumbasierten durchschnittlichen Beziehungsmatrix geschätzt wird, und der Restfehler. Assoziationen mit einem a-log10(P-Wert) größer als fünf wurden als signifikant deklariert.
Ergebnisse
A 1.,5 Mb-Segment auf Chromosom 6 beeinflusst das Laktationsüberleben bei Schweinen
Wir analysierten 31.638 Tiere aus einer einzigen reinrassigen Eberlinie (synthetische Linie mit großem weißen Hintergrund), genotypisiert auf dem Porcine 50K SNP-Chip (Sscrofa11.1 build) (Warr et al., 2019). Die Analyse ergab ein 1,5 Mb-Segment auf Chromosom 6 (SSC6:48,75–50,25), das ein Defizit an Homozygotie zeigte, das mit einem verringerten Laktationsüberleben einherging (Tabellen 1 und 2). Der Haplotyp segregiert bei einer moderaten Allelfrequenz von 4,5% (9,0% Trägerfrequenz) in der untersuchten Population., Die Haplotypfrequenz schwankte in den letzten zehn Jahren, nahm jedoch in den letzten drei Jahren ab (Abbildung S1). Wir haben getestet, ob die Frequenz von einem Vorteilseffekt getrieben wurde. Wir fanden jedoch meist negative Assoziationen mit wichtigen Selektionsmerkmalen mit Ausnahme der Lendentiefe und der Schwangerschaftslänge (Tabelle 3), was darauf hindeutet, dass die Häufigkeit rein auf genetische Drift zurückzuführen ist.

Tabelle 1 SSC6 haplotyp Eigenschaften.,
Tabelle 2 Träger-für-Träger-Würfe zeigen eine Abnahme des Laktationsüberlebens um 24% im Vergleich zu Träger-für-Träger-Würfen. Signifikante Ergebnisse sind fett gedruckt angegeben.

Tabelle 3 Merkmale signifikant assoziiert mit der heterozygoten Träger der SPTBN4 löschen.
Die 52 Träger-für-Träger-Würfe (CxC) zeigen keine signifikante Reduktion der Gesamtzahl der geborenen oder lebendgeborenen Tiere., Das Laktationsüberleben ist jedoch bei CxC-Würfen im Vergleich zu Carrier-by-Noncarrier-Paarungen (CxN) um etwa 24% reduziert, was darauf hinweist, dass homozygote Ferkel innerhalb der Laktationsperiode absterben (Tabelle 2). Als nächstes untersuchten wir die Zeit und die Todesursache von CxC-Würfen. Dies ergab, dass die meisten Ferkel innerhalb der ersten 24 h nach der Geburt starben. Die Mehrheit dieser Ferkel wurde von den Landwirten meist als „schwaches Ferkel bei der Geburt“ beschrieben.,“
Vollgenomsequenzierungsanalyse zeigt eine 16-bp-Frameshift-Deletion in SPTBN4 als wahrscheinliche ursächliche Variante
Um die ursächliche Mutation zu identifizieren, untersuchten wir Ganzgenomsequenzdaten von 71 Tieren aus der untersuchten Population und identifizierten fünf Trägertiere. Die Analyse des Verknüpfungsgleichgewichts (LD) ergab 267 SNP-und Indel-Varianten in hohem LD (r2 > 0.8) mit dem SSC6-Haplotyp (Tabelle S1), wobei die Mehrheit in perfektem LD (247-Varianten) lag., Nur fünf Varianten beeinflussen möglicherweise die Codierungssequenz (drei Missense, ein Frameshift, ein Spleißakzeptor). Es wird vorausgesagt, dass die drei Missense-Varianten von SIFT toleriert werden (score > 0.18, Tabelle S1), während die Splice-Acceptor-Variante ein Gen beeinflusst, das ein 28-BP-Peptid unbekannter Funktion kodiert, wahrscheinlich kausal. Es wurde jedoch vorausgesagt, dass eine Variante des vollständigen LD (r2 = 1) mit dem Haplotyp einen hohen Einfluss hat; eine 16-bp-Frameshift-Deletion in Exon 26 des SPTBN4-Gens (6:g. 48801280delGACGGTGTACGCCGGT) (Abbildungen 1A, B)., Die Frameshift-Deletion (ENSSSCP00000031537:p. Arg1902fs) führt 30 neuartige Aminosäuren und ein vorzeitiges Stop-Codon ein und erzeugt ein gestörtes und abgeschnittenes Spectrin beta non-erythrocytic 4 Protein (SPTBN4). Mutanten fehlen die endgültigen 662 Aminosäuren des Wildtypproteins (Abbildung 1C), einschließlich der Pleckstrin-Homologie (PH) – Domäne, die für den Proteintransport zu Membranen erforderlich ist (Wang et al., 2018). Das SPTBN4-Protein ist ein Mitglied der Beta-Spectrin-Proteine und ein Aktin, das die Zellmembran mit dem Aktin-Zytoskelett verbindet., SPTBN4-Mutationen stören die zytoskeletale Maschinerie, die die ordnungsgemäße Lokalisierung von Ionenkanälen in myelinisierten Nerven steuert und motorische Neuropathien verursacht (Parkinson et al., 2001; Wang et al., 2018).

Abbildung 1 (A) SPTBN4-gen-Modell. Der Ort des betroffenen 26. Exons ist rot markiert. B) Darstellung der 16-bp-Streichung. Abbildung zeigt Wildtyp und mutiertes Exon. (C) Ausrichtung der Mutanten – (Mt) und Wildtyp – (Wt) SPTBN4-Proteinsequenz. Die Mutation induziert 30 neuartige Aminosäuren und ein vorzeitiges Stop-Codon.,
Die Genotypisierung von fünf CxC – Würfen bestätigt die SPTBN4-Deletion als wahrscheinlicher Täter
Wir haben fünf CxC-Würfe für die 16-bp-Deletion genotypisiert, bei der mindestens zwei Ferkel (Bereich 2-6) innerhalb der ersten 48 h nach der Geburt starben. Die fünf Würfe erzeugten 53 Ferkel, von denen 19 für die 16 bp-Deletion homozygot waren (Tabelle 4). Alle 19 homozygoten Ferkel starben innerhalb von 48 h nach der Geburt (18 innerhalb von 24 h). Von den 34 verbleibenden Ferkeln (8 Wildtyp und 26 Träger) starb nur 1 innerhalb von 48 h, wahrscheinlich verursacht durch andere (Umwelt -) Faktoren.,

Tabelle 4 Genotypisierung der wahrscheinlich ursächlichen 16-bp SPTBN4 frameshift-deletion in fünf carrier-by-carrier-Würfe. Die Summe pro Genotypklasse ist fett angegeben.
Homozygote Ferkel für die SPTBN4-Deletion Leiden an Myopathie und Hintergliedparalyse
Wir haben kürzlich einen CxC-Wurf (Abferkeldatum: April 28th 2019) überwacht, der sechs gesunde, zwei betroffene (Proben: 9912, 9916) (Abbildung 2A) und drei totgeborene Ferkel hervorbrachte., Wir bestätigten den homozygoten SPTBN4-Löschstatus für die beiden betroffenen Ferkel (Tabelle S2). Darüber hinaus beobachteten wir vier heterozygote Träger und zwei homozygote Wildferkel unter den gesunden Individuen. Eines der totgeborenen Ferkel (Probe: 9921) war ebenfalls homozygot für die Deletion, während die anderen beiden heterozygot waren. Die betroffenen Ferkel leiden unter extremer Muskelschwäche (Abbildungen 2B, C), Lähmung der Hinterbeine und Zittern (Video S1). Daher konnten die Ferkel nicht laufen oder trinken.,
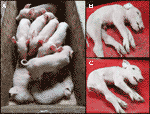
Abbildung 2 (A) die Beiden betroffenen Ferkel (lebendig) zusammen mit sechs gesunden Wurfgeschwister. Die Ferkel stammen aus einer CxC-Paarung, die am 28. B) Befallenes männliches Ferkel 9912. (C) Betroffene weibliche Ferkel 9916.
Betroffenen Ferkeln fehlen Kreuzstreifen in den Skelettmuskeln von Rücken und Hinterbeinen
Die histopathologische Untersuchung ergab eine verstreute Degeneration der Muskelfasern bei beiden Ferkeln und eine fokale Nekrose und Vaskulitis im Rückenmuskel bei einem der Ferkel (ID = 9912)., Darüber hinaus zeigt die Phosphotungstinsäure-Hämatoxylin-Färbung (PTAH) eine divergierende Färbung der Skelettmuskelfasern, was auf eine Abnahme der Querschnitte hinweist, insbesondere in den Muskeln der Rücken-und Hinterbeine der betroffenen Tiere (Abbildung 3B), während die Vorderbeine nicht betroffen zu sein scheinen (Abbildung 3A). Die Abnahme der Kreuzstreifen wird durch abnormale Färbung und allgemeinen Volumenverlust der Muskelfasern angezeigt (Abbildung 3B). Die histopathologisch beobachteten Veränderungen in den Hinterbeinen und in den Rückenmuskeln weisen auf eine Muskeldystrophie hin.,

Abbildung 3 (A) Querschnittsansicht eines Skelettmuskels vom Vorderbein. Der schwarze Pfeil zeigt eine normale Färbung (dunkel) der Muskelfasern an, die auf Kreuzstreifen hinweist. PTAH Bar = 50 µm. (B) Querschnittsansicht eines Skelettmuskels vom Hinterbein. Der schwarze Pfeil zeigt eine abnormale Färbung (rosa) der Muskelfasern an, die auf einen Mangel an Querschnitten hinweist. Der gelbe Pfeil zeigt die normale Färbung und das Vorhandensein von Querschnitten an. PTAH Bar = 50 µm.,
Diskussion
In dieser Arbeit berichten wir über einen neuartigen angeborenen Defekt, der die Ferkelsterblichkeit verursacht, wahrscheinlich aufgrund einer 16-BP-Frameshift-Deletion im SPTBN4-Gen. Die Ferkel leiden unter extremer Muskelschwäche (Myopathie) und sterben innerhalb weniger Stunden nach der Geburt. Es wird erwartet, dass die Deletion einen vollständigen Funktionsverlust des Spektrin-Beta-nicht-erythrozytischen 4-Proteins bewirkt. SPTBN4 ist ein Mitglied der Familie der Spektringene und wird für das Ionenkanalclustering an den Knoten von Ranvier benötigt, was das Aktionspotential beeinflusst (Devaux, 2010)., Mutationen stören die zytoskelettale Maschinerie, die die korrekte Lokalisierung von Ionenkanälen und die Funktion axonaler Domänen hauptsächlich an den Axon-Anfangssegmenten (AIS) und den Ranvier-Knoten steuert (Wang et al., 2018). Genauer gesagt ist die betroffene C-Terminal-Domäne von SPTBN4 entscheidend für den KCNQ2-Kanalhandel und die Erregbarkeit an Ranvier-Knoten (Devaux, 2010).
Nachfolgende Follow-up-Untersuchungen identifizierten Fälle von Mensch und Maus, die darauf hindeuteten, dass sich das folgende Syndrom wahrscheinlich nicht sofort als tödlich erweisen würde, sondern eine schwere Myopathie hervorrufen würde., Durch SNP-Genotypdaten mittlerer Dichte, die für alle Tiere in der Zuchtpopulation verfügbar sind (N = 31,839), konnten Träger identifiziert werden. Unter diesen Trägern befand sich eine Sau, die zum Zeitpunkt der Identifizierung ungefähr zur Halbzeit der Schwangerschaft war und von einem Eber gezeugt wurde, der ebenfalls Träger war. Der Zuchtbetrieb wurde benachrichtigt, um den Wurf bei der Geburt zu dokumentieren. Der beobachtete Phänotyp der betroffenen Ferkel (Myopathie, Lähmung der Hintergliedmaßen, Zittern) stimmte vollständig mit dem überein, was bei menschlichen Patienten mit homozygot Funktionsverlust oder zusammengesetzten heterozygoten Mutationen im SPTBN4-Gen beobachtet wurde (OMIM: 606214)., Zwei der menschlichen Patienten haben Funktionsverlustmutationen innerhalb der PH-Domäne (Wang et al., 2018), dass ein Verlust der PH-Domäne bei Schweinen wahrscheinlich zu einem vollständigen Funktionsverlust des SPTBN4-Proteins führen würde. Beim Menschen führen ähnliche Mutationen zu einer schweren kongenitalen Myopathie, die durch das Fehlen von Muskelfasern vom Typ I, Neuropathie und Taubheit verursacht wird (Knierim et al., 2017; Wang et al., 2018). Wang et al. (2018) beobachtete auch motorische axonale Neuropathie bei mehreren Patienten, die durch angeborene Hypotonie, tiefe Schwäche und Verlust tiefer Sehnenreflexe in der frühen Kindheit gekennzeichnet waren., Darüber hinaus zeigten Nervenbiopsien reduzierte nodale Na+ – Kanäle und keine nodalen KCNQ2 K+-Kanäle, was die molekulare Pathologie aufdeckte, die eine Funktionsstörung des Nervensystems verursachte. Daher schließen wir, dass diese Frameshift-Variante die wahrscheinliche ursächliche Mutation ist, die zum beobachteten Phänotyp und zur Erschöpfung des homozygoten Genotyps in der Population führt. Zukünftige Studien könnten sich darauf konzentrieren, ein In-vivo-Knockout des SPTBN4-Gens bei Schweinen durchzuführen, um das Syndrom und den damit verbundenen Phänotyp genauer zu untersuchen.,
Wir haben keine Degeneration von Muskelfasern in den Vorderbeinen beobachtet, während die dorsalen und Hinterbeinmuskelfasern deutlich betroffen waren. Diese Beobachtung könnte teilweise die Lähmung der Hinterbeine erklären, während die Vorderbeine nicht betroffen sind. Die Diskrepanz zwischen Vorder-und Hinterbeinmuskelfasern wurde auch bei zitternden Mäusen beschrieben, bei denen SPTBN4-Funktionsverlustmutationen Motoneuropathie, Lähmung der Hinterbeine, Zittern und zentrale Taubheit verursachen (Parkinson et al., 2001; Komada und Soriano, 2002). Parkinson et al., (2001) Beschreiben Sie reduzierte Nervenleitungsgeschwindigkeiten in Ischiasnerven von Mäusen mit zitternden Allelen, die die periphere Neuropathie der Hintergliedmaßen verursachen. Die Expression von SPTBN4 bei Mäusen ist auf das Gehirn, das Rückenmark und die Ischiasnerven beschränkt und wird im Skelettmuskel nicht beobachtet, so dass diese Krankheit in erster Linie ein neuronaler Defekt ist. Insgesamt bleibt unklar, welcher Mechanismus das Fehlen von Symptomen in den Vorderbeinen verursacht. Dieser „natürliche Knockout“ bei Schweinen kann eine nützliche Ressource sein, um die menschliche Krankheit zu untersuchen, da Schweine im Vergleich zu Nagetierarten normalerweise ein besseres Modell zur Untersuchung menschlicher Krankheiten sind., Darüber hinaus kann die Folge des Verlustes der SPTBN4-Funktion genauer untersucht werden.
Die effektive Populationsgröße (Ne) der untersuchten Rasse wird auf rund 100 geschätzt (Hidalgo et al., 2016). In der Tierzucht erhöht ein niedriger Ne das Risiko, dass schädliche Allele zufällig häufiger auftreten. Darüber hinaus haben frühere Studien gezeigt, dass rezessive letale Allele durch vorteilhafte Wirkungen bei Heterozygoten angetrieben werden können (Derks et al., 2018; Matika et al., 2019). Matika et al.,, 2019 fand eine rezessive Stop-Stop-Mutation im MSTN-Gen, die mit einer starken Zunahme der Muskeltiefe bei Heterozygoten assoziiert war. Wir finden jedoch keine Beweise für einen heterozygoten Vorteil in unserer Studie. Mit den aktuellen genomischen Techniken können wir nun schädliche Allele identifizieren, die zu höheren Frequenzen driften, und die Entstehung neuartiger schädlicher Allele genau überwachen, was eine effektivere Reinigung ermöglicht. Darüber hinaus wird das Ergebnis dieser Art von Studie das Bewusstsein für „versteckte“ genetische Defekte sowohl auf Züchter-als auch auf Bauernebene erheblich verbessern., Ohne vorherige Information werden seltene Geburtsfehler oft als „schwaches Ferkel“ registriert.“Und ohne weitere Unterscheidung spezifischer Syndrome sind weitere Maßnahmen nicht möglich. In den meisten Fällen ist nicht bekannt, ob es eine genetische Grundlage gibt oder ob es andere verwirrende Wirkungen geben kann. Mit früheren genomischen Informationen kann das Syndrom im Vergleich zu anderen Fällen identifiziert und Träger identifiziert werden, was zu umsetzbaren Informationen führt.
Die Ferkelsterblichkeit ist von hoher wirtschaftlicher und tierschutzpolitischer Bedeutung., Daher hat die Entdeckung der SPTBN4-Mutation zu einer sofortigen Implementierung in das Zuchtprogramm geführt, um die Häufigkeit von Träger-für-Träger-Paarungen zu minimieren. Dies ermöglicht es, die Geburt betroffener Personen zu vermeiden, wodurch der Tierschutz verbessert und wirtschaftliche Verluste verringert werden.
Schlussfolgerung
In dieser Studie berichten wir über einen neuartigen angeborenen Defekt, der wahrscheinlich durch eine rezessive Frameshift-Deletion im SPTBN4-Gen bei Schweinen verursacht wird. Die Ergebnisse werden durch auffallende Ähnlichkeiten mit SPTBN4-assoziierten Syndromenphänotypen bei Menschen und Mäusen gestützt., Die Studie ermöglicht es, das schädliche Allel aus der Bevölkerung zu überwachen und zu reinigen. Träger-für-Träger-Kreuze können verhindert werden, indem betroffene Personen ausgeschlossen werden, wodurch wirtschaftliche Verluste verringert und der Tierschutz verbessert wird. Schließlich können diese „natürlichen Knockouts“, die in der Zuchtindustrie erhalten werden, ein Modell für menschliche Krankheiten liefern und unser Verständnis der Genfunktion innerhalb der Säugetierklate verbessern und ein potenzielles Modell für menschliche Krankheiten liefern.,
Verfügbarkeit der Daten-Anweisung
Ethik-Statement
die Ethische überprüfung und Genehmigung war nicht erforderlich, für die Tier-Studie, da die Daten in dieser Studie verwendet wurde, erhalten als Teil der routinemäßigen Sammlung von Daten von Topigs Norsvin der Zucht, und nicht speziell für den Zweck dieses Projekts. Daher war die Zustimmung einer Ethikkommission nicht zwingend erforderlich. Die Probenentnahme und Datenerfassung erfolgte streng nach dem niederländischen Tierschutzgesetz (Gezondheids-en welzijnswet voor dieren)., Die schriftliche Einwilligung der Besitzer zur Teilnahme ihrer Tiere an dieser Studie wurde von den Besitzern eingeholt.
Autorenbeiträge
MG, H-JM und MD konzipierten und gestalteten die Studie. BH war für die allgemeine Organisation und Kommunikation mit Topigs Norsvin und Bauern verantwortlich. MD und ML führten die Datenanalyse durch. BD und KL führten Laborarbeiten durch. SG-V führte die pathologische Analyse durch. MD schrieb das Manuskript. H-JM, MG, BH, SG-V, BD, KL und ML lieferten nützliche Kommentare und Vorschläge und halfen beim Entwurf des Manuskripts. Phänotypische Daten wurden von ML analysiert., Alle Autoren lasen und genehmigten das endgültige Manuskript.
Finanzierung
Diese Forschung wurde finanziert von der STW-Breed4Food Partnerschaft, Projekt-Nummer 14283: Von der Sequenz zum Phänotyp: die Erkennung schädlichen variation durch Vorhersage der Funktionalität. Diese Studie wurde von NWO-TTW und den Breed4Food Partnern Cobb Europe, CRV, Hendrix Genetics und Topigs Norsvin finanziell unterstützt. Darüber hinaus wurde diese Studie durch das IMAGE-Projekt (Horizon 2020, Nr. Die Geldgeber hatten keine Rolle in Studiendesign, Datenerfassung und-Analyse, der Entscheidung zur Veröffentlichung oder der Vorbereitung des Manuskripts., Die Nutzung des HPC-Clusters wurde durch CATAgroFood (Shared Research Facilities Wageningen UR) ermöglicht.
Haftungsausschluss
Die in dieser Studie verwendeten Daten wurden im Rahmen der routinemäßigen Datenerfassung von Topigs Norsvin-Zuchtprogrammen und nicht speziell für den Zweck dieses Projekts erhalten. Daher war die Zustimmung einer Ethikkommission nicht zwingend erforderlich. Die Probenentnahme und Datenerfassung erfolgte streng nach dem niederländischen Tierschutzgesetz (Gezondheids-en welzijnswet voor dieren).,
Interessenkonflikt
ML und BH sind Mitarbeiter des Topigs Norsvin Research Center, eines Forschungsinstituts, das eng mit einem der Geldgeber (Topigs Norsvin) verwandt ist.
Alle verbleibenden Autoren erklären, dass die Forschung ohne kommerzielle oder finanzielle Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Die anderen Breed4Food-Partner Cobb Europe, CRV und Hendrix Genetics erklären, für diese Studie keine konkurrierenden Interessen zu haben.,
Danksagungen
Die Autoren danken Frank van Haaren und Toon Janssen für die Organisation der Kommunikation mit der Farm. Wir danken Manon Houben für ihren Beitrag zu den pathologischen Untersuchungen. Wir danken Mout Viller für die Aufnahme von Videos und Bildern der betroffenen und gesunden Ferkel nach der Geburt. Wir danken Jürgen Harlizius für die Betreuung der Ferkel nach der Übergabe. Wir danken Gerda van Eldik für die Probenentnahme. Wir danken Egbert Knol und Egiel Hanenberg für die allgemeine Unterstützung und Anleitung von Topigs-Norsvin.,
Ergänzungsmaterial
Video S1 / Video zeigt die beiden betroffenen Personen nach der Geburt.
Tabelle S1 / Genomische Variation in hohem LD mit dem Haplotyp SSC6.
Tabelle S2 / Genotypisierung des CxC-Wurfes mit zwei betroffenen Individuen (Abferkeldatum: 28. Tabelle zeigt homozygoten del / del-Status für die beiden betroffenen Ferkel 9912,9916.
Tabelle S3 / Mapping-und Abdeckungsstatistiken aus WGS-Stichproben.
Abbildung S1 | SPTBN4 Löschung Trägerfrequenz von 2007-2019.