Das Gen p53 wurde erstmals 1979 entdeckt. Ein Protein wurde in Simian Virus 40-transformierten Mauszellen (SV40) durch Immunpräzipitation mit Anti-T-Serum identifiziert; Dieses Protein wurde Protein p53 genannt . Im selben Jahr fanden Kress und andere Wissenschaftler eine neue Klasse von Proteinen mit einer molekularen Masse von 50-60kDa. Diese Art von Protein wurde dann als p53 identifiziert . Das Protein p53 kann auch aus verschiedenen transformierten Zelllinien durch Immunpräzipitation identifiziert werden., Lane und Linzer erzielten 1979 ebenfalls ein ähnliches Ergebnis. Andere Beweise für die Identifizierung von p53 sind, dass p53 in allen getesteten transformierten Mauszellen exprimiert wurde; Diese Tests umfassen chemisch induzierte Sarkome, transformierte Fibroblasten und Leukämien, während in normalen Zellen p53 nicht exprimiert wurde. Darüber hinaus wurde in den meisten transformierten Zellen ein hoher p53-Spiegel nachgewiesen, unabhängig davon, wie die Zellen entweder spontan oder nicht spontan transformiert wurden ., Dies war weitgehend auf die erhöhte Stabilität des p53 zurückzuführen, exprimierte jedoch in F9-embryonalkarzinogenen Zellen einen hohen p53-Spiegel, was auf die Menge der translated p53-mRNA zurückzuführen war .
Nachdem das Protein p53 1979 entdeckt worden war, wurde es populär, es zu analysieren. Da es sich jedoch um ein neu entdecktes Protein handelte und es keinen früheren Namen dafür gab, verwendeten verschiedene Institutionen unterschiedliche Namen und veröffentlichten Artikel mit unterschiedlichen Namen., Um dieses Problem zu lösen, trafen sich 1983 während des 1.Internationalen p53-Workshops in Oxted, Großbritannien, Wissenschaftler verschiedener Forschungsgruppen in verschiedenen Ländern, um eine gemeinsame Nomenklatur für dieses neu entdeckte Protein zu diskutieren. Bei diesem Treffen wurde “ p53 “ zu seinem Namen und wird seitdem verwendet. Es wurde angenommen, dass der Grund, warum Wissenschaftler das Protein p53 nannten, darin besteht, dass die molekulare Masse dieses Proteins 53kDa ist, die auf seiner Migration in SDS-Gel basiert. Später wurde die molekulare Masse als falsch erwiesen, und die richtige molekulare Masse sollte 43 sein.,7kDa, weil p53 eine prolinreiche Region enthält und diese Region die Migration von p53 in SDS-Gel reduzieren kann. Aber der name „p53“ blieb .
In den 1980er Jahren wurde angenommen, dass das Protein p53 am Zellzyklus beteiligt ist und eine Rolle bei der DNA-Replikation spielt. Später, in den Jahren 1982 bis 1994, stellten die Menschen fest, dass einige virale Onkoproteine an p53 binden und einen Komplex bilden konnten. In 1982, Sarnow et al. es wurde festgestellt, dass Adenovirus E1b (58kDa) mit einem 54kDa-Protein interagieren kann, das in den oben genannten SV40-transformierten Mauszellen vorhanden ist., Nach den Ergebnissen der immunologischen Spezifitäten von T-Antikörpern und den Peptidkarten des 54kDa-Proteins wird dieses 54kDa-Protein als p53 identifiziert . Im selben Jahr fanden Wissenschaftler heraus, dass, wenn sie den p53-Antikörper in Schweizer 3T3-Mauszellen injizieren würden, dies Zellen hemmen würde, die in die S-Phase des Zellzyklus eintreten; Unter der gleichen Situation beeinflusste der p53-Antikörper die SV40-oder Adenovirus-induzierte DNA-Synthese nicht .,
Später im Jahr 1984 untersuchten Wissenschaftler die Wirkung des p53 auf nicht transformierte 3T3-Fibroblasten; Sie analysierten die Syntheserate des Proteins p53 zu verschiedenen Zeitpunkten und fanden heraus, dass in der späten G1-Phase die Syntheserate und das Niveau des Proteins p53 und seiner verwandten mRNA zunehmen. Dieses Ergebnis legt nahe, dass das Protein p53 Zellen hemmt, die aus der Interphase in die Teilungsphase eintreten . Maltzman W et al. habe im selben Jahr ein weiteres Experiment gemacht. Sie behandelten die nicht transformierte Mauszelle mit UV-Licht und UV-mimetischem chemischem Karzinogen 4NQO und stellten einen hohen p53-Spiegel fest., Das Ergebnis zeigte, dass die erhöhte Expression von p53 nicht nur ein Symbol ist, das den Zellzyklus anzeigt, sondern vor allem auch eine Komponente, die an der DNA-Synthese und Zellproliferation beteiligt ist . Bei der Untersuchung des Komplexes des T-Antigens des Simian-Virus 40 und der DNA-Polymerase α fanden Gannon und andere Wissenschaftler 1987 eine ähnliche Veränderung des Antigens, wenn es an p53 und Polymerase α gebunden war. Sie fanden auch heraus, dass sie bei einer bestimmten Konzentration der drei Komponenten einen speziellen trimeren Komplex bilden können, der T-Antigen, p53 und DNA-Polymerase α enthält., Da T-Antigen an der viralen DNA-Replikation und zellulären Transformation beteiligt ist, zeigt dieses Ergebnis, dass p53 eine Rolle bei der Kontrolle des Zellzyklus und der DNA-Replikation spielt .
Wie das Experiment oben gezeigt hat, hat p53 die Fähigkeit, Zellen zu verewigen. In 1984, Eliyahu D et al. gefunden, dass p53 und das Produkt von Oncogene myc einige ähnliche Eigenschaften teilten. Beide haben die Fähigkeit, an andere Proteine zu binden und sind am Zellzyklus beteiligt, und beide akkumulieren in Kernen transformierter Zellen ., Bienz, Pennica und Oren analysierten die Aminosäuresequenzen des Proteins p53 und des Produkts von myc und stellten fest, dass die beiden Proteine Ähnlichkeiten in der molekularen Struktur und der Position von speziell geladenen Resten aufweisen. Dann schlugen Wissenschaftler vor, dass p53 als Onkogen wirken könnte. Basierend auf dieser Hypothese, Eliyahu D et al. hat einige Experimente gemacht. Da die primären Rate Embryo Fibroblasten durch die Beteiligung von myc Produkt und Ha-ras transformiert werden können, primäre Baby Ratte Nierenzellen können auch durch die Zusammenarbeit von Ha-ras und Adenovirus frühen Region 1A transformiert werden, Eliyahu D et al., beschlossen, diese Art von biologischem Testsystem zu verwenden, um die onkogene Funktion des p53 zu identifizieren. Sie behandelten normale embryonale Zellen mit p53 und aktivierten Ha-ras. Das Ergebnis zeigte, dass Zielzellen auf morphologische Veränderungen stoßen und hohe p53-Spiegel produzieren, Eliyahu D et al. dachte, dass die Transformation von embryonalen Fibroblasten durch p53 und Ha-ras erklärte, dass das Gen p53 ein Onkogen ist ., 1985 schlug Jenkins vor, dass das p53-Gen die Lebensdauer von Zellen verlängern und die Affektivität der Transformation verbessern kann, indem es seine kodierende Sequenz neu anordnet, die die Produktion stabiler Proteine verursachen könnte .
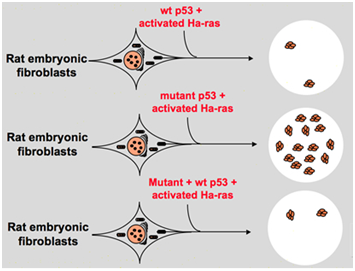
In den späten 1980er Jahren erkannten Wissenschaftler jedoch, dass p53 ein Tumorsuppressorgen anstelle eines Onkogens ist. Sie beobachteten, dass p53 mit normaler Funktion in vielen Tumoren nicht nachgewiesen werden kann, und stellten fest, dass der Verlust der Expression und Funktion des Wildtyp-p53-Gens während der Zelltransformation notwendig ist., Diese erhöhen die Möglichkeit, dass Wildtyp – p53-Gen neoplastische Progression hemmen kann . Dann formulierten sie eine andere Hypothese: Das in früheren Experimenten verwendete Klongen p53 enthält gelegentlich dominante negative Mutationen innerhalb der hochkonservierten Domäne, die zu entgegengesetzten Experimentergebnissen führen . Im Jahr 1988 entdeckten Ben und andere Wissenschaftler eine riesige Menge an neu angeordnetem p53 in murinen Erythroleukämie-Zelllinien-DP20-1 und CB3, die von den Milben der mit dem Freund-Leukämievirus infizierten Murine abgeleitet sind ., 1989 änderte Eliyahu, der darauf hinwies, dass p53 ein Onkogen ist, seine Meinung und nahm an, dass das Wildtyp-p53-Gen die Zelltransformation hemmen könnte. Eliyahu und andere Wissenschaftler untersuchten die Wirkung von Wildtyp – p53-Protein, das von Plasmiden und mutiertem p53 kodiert wird, auf die Fähigkeit, die primäre Embryo-Fibroblast-Transformation durch verschiedene Onkogenkombinationen in vitro zu bewirken. Zum Beispiel mutant p53 plus ras und myc plus ras., Das Ergebnis zeigte, dass Wildtyp-p53 zu einer enormen Verringerung der transformierten Herde führen, die durch mutierte p53 plus ras verursacht werden; Mutierte p53 zeigten keine Hemmung transformierter Herde, die durch myc plus ras verursacht wurden, während myc plus ras-vermittelte Transformation sehr empfindlich auf die Expression von Wildtyp-p53 reagiert. Abbildung 1 zeigt dieses Experiment prägnant. Es zeigte sich, dass Wildtyp p53 im Vergleich zu mutiertem p53 eine offensichtlich hemmende Wirkung auf die Zelltransformation aufweist. Der Effekt hängt positiv mit dem Expressionsniveau des Wildtyps p53 und negativ mit dem Expressionsniveau der mutierten p53 zusammen., Dieses Experiment deutete darauf hin, dass Wildtyp p53 im Vergleich zu mutiertem p53 tatsächlich eine entgegengesetzte Funktion haben und die Tumorigenese hemmen kann . Derzeit wird p53 als Tumorsuppressor-Gen erkannt. Es wird geschätzt, dass etwa die Hälfte der Tumoren durch p53 verursacht wird. Es ist eines der am häufigsten mutierten Gene beim Menschen und das weltweit am häufigsten analysierte Gen .
In den ersten Jahren der 1980er Jahre waren der biochemische Weg von p53 und die Wirkung der p53-Mutation nicht klar. Im Jahr 1991 fanden Kern und andere Wissenschaftler heraus, dass eine 33-Basen-Paar-DNA-Sequenz spezifisch an Wildtyp-p53 in vitro bindet. Sie fanden auch heraus, dass das p53-Protein zwei Mutationen enthält, die normalerweise in menschlichen Tumoren vorkommen, die nicht an diese spezifische DNA-Region binden können. Sie vermuten also, dass die Funktion von p53 von seiner Fähigkeit abhängt, spezifische DNA-Sequenzen zu binden, und diese Fähigkeit wird durch Mutationen in menschlichen Tumoren verändert., Sie nehmen auch an, dass diese 33-Basen-Paar-DNA-Sequenz möglicherweise nicht die einzige Sequenz ist, die die Fähigkeit hat, spezifisch an das p53 beim Menschen zu binden; Es kann jedoch Menschen helfen, die Funktion von p53 besser zu verstehen . Später wurde festgestellt, dass p53 während des Zellzyklus, der DNA-Reparatur, der Differenzierung, der Initiierung von Apoptose und Angiogenese eine Rolle spielt. Rotter V et al. gefunden, dass p53 up – reguliert die Differenzierung von Zellen. Beispielsweise wurde in mehreren Schlüsselschritten während der B-Zell-Differenzierung ein hoher Gehalt an p53-Protein nachgewiesen. Erhöhtes p53 kann auch während der Spermatogenese nachgewiesen werden., Inzwischen kann in einigen Organen erwachsener Mäuse nur ein sehr niedriger p53-Proteingehalt nachgewiesen werden .
1990 wurde gelegentlich ein nützliches Werkzeug entdeckt. Es ist eine temperaturempfindliche Mutante von p53, genannt p53val135. Es kann als echter Wildtyp p53 bei der Temperatur von 32,5 oC wirken und die Transformation unterdrücken, und es kann auch als anderer mutierter p53 bei der Temperatur von 37,5 oC oder über 48oC wirken und die Transformation auslösen. Zusätzlich wird für transformierte Zellen, die p53val135 exprimieren, seine Proliferation bei der permissiven Temperatur kontrolliert, und diese Art der Kontrolle ist reversibel., Durch die Verwendung dieser p53val135-Mutante wurde Wildtyp-p53 entdeckt, um einen Zellzyklusstillstand bei G1 oder G2/M zu induzieren . Im Jahr 1991 Elisheva et al. es wurde festgestellt, dass das temperaturempfindliche p53val135 eine andere Funktion in der murinen myeloischen Leukämiezelllinie ausübte. Nach der Reaktivierung von p53val135 für einige Tage starben alle Zellen, und dieser Tod zeigt einige Eigenschaften der Apoptose . Ein Jahr später erhielt Shaw ein ähnliches Ergebnis. Ein Wildtyp p53 wurde in eine humane Kolontumor-abgeleitete Zelllinie EB transferiert., Die Zellen wurden unter Licht-und Elektronenmikroskopen untersucht und zeigten einige Eigenschaften der Apoptose . In 1990, Scheffner et al. und andere Wissenschaftler fanden heraus, dass E6, das die Zerstörung von regulatorischen Proteinen der Wirtszellen stimuliert, von den onkogenen humanen Papillomavirus-Typen 16 und 18 kodiert wird und einen Komplex mit Wildtyp-p53 in vitro bilden kann, der wiederum den Abbau von Protein-p53 verursacht .
1992 wurde ein Schlüsselprotein MDM2 entdeckt, weil es fest an p53 bindet und die durch p53 vermittelte Transaktivierung hemmt., Die molekulare Masse von MDM2 beträgt 90kDa und bildet einen Komplex mit mutiertem und Wildtyp-p53 . Im selben Jahr, Livingstone RL et al. untersucht, ob die Zelle eine oder beide Kopien von Wildtyp-p53-Allelen verloren hat und ob dies ausreicht, um eine Genamplifikation zu verursachen. Die Genamplifikation wurde hauptsächlich in transformierten Zellen nachgewiesen, jedoch nicht in den normalen Fibroblasten. Das Ergebnis zeigte, dass Zellen, die eine Kopie der p53-Allele verlieren, als Wildtyp-p53 wirken, während Zellen, die beide Kopien der Wildtyp-p53-Allele verlieren, eine höhere Amplifikationsfrequenz aufweisen . Ein weiteres Experiment von Yin Y et al., zeigte ein ähnliches Ergebnis .
1993 wurde ein p53-Zielgen namens CDKN1A identifiziert. Es kodiert das Protein p21, das ein Cyclin-abhängiger Kinase-Inhibitor ist, der Cyclin-CDK2 und CDK1 durch Bindung an sie hemmt. 1993 fand Szekely heraus, dass das Epstein-Barr-Virus-Kernantigen 5(EBNA-5) durch das Epstein-Barr-Virus kodiert ist und humane B-Lymphoblastoidzellen infizieren kann. Ein 66 Aminosäure langes Peptid ist für die Bildung von komplexem EBNA-5-p53 verantwortlich, Punktmutationen von p53 beeinflussten nicht seine Bindungsfähigkeit zu EBNA-5., Es hemmt jedoch seine Komplexbildung mit anderen Molekülen . 1994 beschrieben Cho und seine Mitarbeiter erstmals die Kristallstruktur komplexer p53-DNA. Diese DNA-Bindungsdomäne wurde auch als Kerndomäne bezeichnet. Es enthält Rückstände 102-292, und besteht aus einem beta-sandwich. Sie demonstrierten auch die detaillierte Struktur der Kerndomäne . Auch im Jahr 1994, Wang XW et al. die Interaktion zwischen hepatitis-B-virus-X-protein (HBX) und Wildtyp-p53-protein in Menschen., Sie fanden heraus, dass HBX die Fähigkeit von p53 hemmen kann, an andere sequenzspezifische DNA zu binden, nachdem es an p53 gebunden ist, und es kann auch die Assoziation von p53 mit Transkriptionsfaktoren hemmen .
1997 Honda R et al. zunächst wurde die Hypothese aufgestellt, dass MDM2 die Ubiquitylierung von p53 auslösen und zum Abbau von p53 durch ein Ubiquitin-Proteasom-System führen kann. Sie wiesen darauf hin, dass MDM2 an die N-terminale Domäne (NTD) von p53 bindet und als Ubiquitinligase E3 wirkt . Ebenfalls 1997 wurden zwei neue Proteinfamilien, p63 und p73, entdeckt, die eine wesentliche Homologie mit p53 teilen., p73, auch Tumorprotein 73 genannt, wird von einem Gen in 1p36 kodiert. Der Ort wird häufig bei Neuroblastomen und anderen Tumoren gelöscht. p73 kann p53-Zielgene aktivieren und interagiert mit p53 . Yang et al. es wurde festgestellt, dass sich das Gen p63 in 3q27-29 befindet und in verschiedenen Maus-und menschlichen Zellen nachgewiesen werden kann. Wie p73 kann p63 p53-Zielgene signifikant transaktivieren, es kann auch Apoptose induzieren. Ein Merkmal von p63 ist, dass die Mehrheit von p63 keinen N-Terminus hat ., Im selben Jahr fanden Serrano und Kollegen heraus, dass primäre murine Fibroblasten durch ras in Abwesenheit von p53 oder p16 transformiert werden können und inaktives p53 oder p16 den Verewigungsprozess menschlicher Zellen erleichtern kann. Diese Ergebnisse legen nahe, dass p53 eine Rolle bei der zellulären Seneszenz spielt . 1997 wurde festgestellt, dass p53 eine Rolle bei der Initiierung der Apoptose spielt. Wenn Zellen in die Proliferationsphase kommen, verkürzen sich die Telomere am Ende jedes Chromosoms nach jeder Runde der DNA-Replikation aufgrund unvollständiger Replikation der Einzelstrang-DNA am Ende des DNA-Ständers ., Aktiviertes Tumorsuppressor-Gen p53 begrenzt die Häufigkeit, mit der eine Zellteilung auftreten kann. Wynford TD fand heraus, dass mit dem Verlust der Funktion des Wildtyps p53 alle Fibroblasten aus der Apoptose entkommen. Auch die Transaktivierungsfunktion von p53 kann durch Apoptose aktiviert werden . Wynford TD schlug vor, dass es drei Möglichkeiten gibt, wie p53 aktiviert wird. Die erste ist die posttranslationale Modifikation durch Phosphorylierung, die zweite ist die Hochregulierung der transkritionalen Cofaktoren wie p33ING1, die letzte ist die Herunterregulierung der p53-Inhibitoren wie MDM2 .
Brodsky MH et al., studierte die Transkriptionsziele von p53 in Drosophila. Es gibt Hinweise darauf, dass Drosophila-Augen einen schweren groben Augenphänotyp unter der Expression von menschlichem p53 aufweisen, der Apoptose von imaginären Augenscheibenzellen induziert, den Verlust von Pigmentzellen verursacht und schließlich die Entwicklung von Drosophila hemmt Drosophila kann also ein Modelltier sein , um die Funktion von p53 zu untersuchen. Brodsky fand heraus, dass das Gen rpr eine Konsensus-p53-Bindungsstelle enthält, die sich in der cis-regulatorischen Region von rpr befindet, und es ist auch ein Aktivator der Apoptose., Mit anderen Beweisen behauptete Brodsky, dass rpr ein Transkriptionsziel des p53 ist . Im Jahr 2001 fanden Derry und Kollegen heraus, dass C. elegans kein p53-Gen haben, sondern tatsächlich ein Gen cep-1 enthalten, das Proteine kodiert, die eine ähnliche Sequenz mit Protein p53 haben. Dieses C. elegans-Gen kodiert für Protein CEP-1, das die Fähigkeit hat, Apoptose durch genotoxischen Stress zu induzieren, und ist eine notwendige Komponente während der Meiose .
Im Jahr 2002 schlugen Tyner und Kollegen vor, dass p53 eine Rolle bei der Regulierung der Alterung von Organismen spielt., Um die Funktion von p53 zu untersuchen, schufen sie gentechnisch veränderte Mäuse mit mutiertem p53, indem sie Exons 1-6 und eine stromaufwärts liegende Region des Wildtyp-p53-Gens (p53+/+) löschten, genannt p53+/m. Es wirkt als Wildtyp-p53 und hat eine verbesserte Resistenz gegen spontane Tumoren besser als Wildtyp-p53. Im Experiment überwachten sie die Mäuse, die p53+/m, p53+/+ und p53+/-enthielten. p53+ / – bedeutet, dass die Mäuse eine Kopie des Wildtyps p53-Gens verlieren., Die Ergebnisse zeigten, dass keiner der Mäuse mit p53+/m entwickelt lebensbedrohlichen Tumoren, jedoch mehr als 80% der Mäuse mit p53+/- und mehr als 45% der Mäuse mit p53+/+ entwickelt diese Arten von Tumoren. In 2 von 35 p53+/m-Mäusen wurden lokalisierte Tumorläsionen beobachtet, im Gegensatz dazu wurden verschiedene Tumoren wie Lymphome und Osteosarkome in p53+/- und p53+/+ – Mäusen gefunden. Während dieses Experiments beobachteten sie auch, dass das Durchschnittsalter von p53+/m 96 Wochen betrug, während das Durchschnittsalter von p53+/m 116 bis 118 Wochen betrug., Tyner und Kollegen untersuchten auch die Möglichkeit, dass die kürzere Lebensdauer von p53+/m mit dem Altern verbunden war. Sie fanden heraus, dass nach 18 Monaten die p53+/m-Mäuse begannen, Gewicht und Kraft zu verlieren, wie bei p53+/m-Mäusen wurden im Alter von 30-36 Monaten reduzierte Gewichte beobachtet. p53+ / m-Mäuse zeigen auch Lordokyphose. Abhängig von der Röntgenanalyse zeigten p53+/m-Mäuse im Alter von 12 Monaten eine verringerte Knochendichte und werden im Alter von 18 Monaten schwerwiegend. Dies ist ein Symbol für Osteoporose und Osteoporose ist ein Marker für das Altern bei Menschen und Mäusen . Tyner et al., getestet wurde auch die Toleranz von Stress, da diese Fähigkeit auch ein Marker für das Altern ist . Sie führten 3 mm Stanzbiopsien in der Rückenhaut von alten und jungen narkotisierten p53+/m-und p53+/+ – Mäusen durch. Ihre Ergebnisse zeigten, dass viele alte p53+/m-Mäuse nach der Injektion von Standarddosis Avertin starben, was darauf hindeutet, dass alte p53+/m-Mäuse weniger tolerant gegenüber Stress waren .
Im Jahr 1991 wurde festgestellt, dass p53 die Fähigkeit hat, Apoptose zu induzieren, während im Jahr 2003; Mihara und andere Wissenschaftler fanden heraus, dass p53 auch eine Apoptose Rolle in den Mitochondrien hat ., Da einige mitochondriale Proteine die Fähigkeit haben, die zelluläre Apoptose entweder durch aktive Caspasen oder durch neutralisierende cytosolische Inhibitoren zu aktivieren. Im Beispiel der Cytochrom-c-induzierten Caspase wird nach Empfang des Apoptosesignals Cytochrom c aus dem Intermembranraum der Mitochondrien freigesetzt und bindet dann wiederum an Apf-1, das als inaktives Monomer existiert, seine Konformationsänderung induziert und seine Bindungsaffinität für dATP/ATP um das Zehnfache erhöht als Apaf-1 allein dATP/ATP bindet. Dann bindet das komplexe Apaf-1-Cytochrom c an dATP / ATP und bildet das Apoptosom., Danach wird die Caspase-Rekrutierungsdomäne (KARTE) von Apaf-1 im Apoptosom exponiert, rekrutiert Procaspase-9 und aktiviert sich dann selbst. Der endgültige Komplex spaltet und aktiviert dann andere Caspasen wie Caspase-3, die wiederum wichtige Moleküle in der Zelle spalten, was zu Chromatinkondensation, DNA-Fragmentierung und schließlich zu Apoptose führt . Abbildung 2 zeigt den Cytochrom-c-induzierten Caspase-Aktivierungsweg.
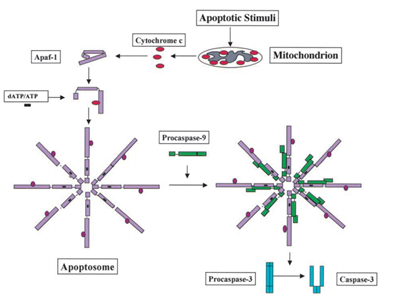
Mihara M et al. wissenschaftler fanden heraus, dass das Wildtyp-p53-Gen schnell in die mitochondriale Oberfläche von Tumorzellen transloziert werden kann. In dem Experiment fanden sie heraus, dass ein stressinduziertes Wildtyp-p53-Protein die Fähigkeit hat, nach Apoptose aufgrund von DNA-Schäden und Hypoxie in die Mitochondrien von Thymozyten in menschlichen oder Mauszellen zu translozieren. Dann induzieren diese Wildtyp-p53-Proteine die Permeabilisierung von Mitochondrien und verursachen eine Reihe von Veränderungen, die in Mitochondrien auftreten, wie die Freisetzung von Cytochrom c durch Bildung eines Komplexes mit Bcl2 und BclXL .,
Als gutes klinisches Ergebnis mit geringer Nebenwirkung ist die Gentherapie beliebt. Bis Ende 2005 gab es 1020 Gentherapiestudien in der Datenbank des Journal of Gene Medicine. Unter diesen Studien wurden 66% der Gentherapien an Krebspatienten durchgeführt, und 58 Studien verwendeten rAd-p53, ein rekombinantes Adenovirus, das für das humane p53-Gen kodiert. Im April 2004 wurde eine rekombinante humane Adenovirus-p53-Injektion (Gendicin) offiziell eingeführt. Gendicine wird zur Behandlung von Kopf-Hals-Plattenepithelkarzinom und es wurde von der staatlichen Food and Drug Administration of China am Okt. 16, 2003., Es wurde das erste Gentherapieprodukt der Welt, das von der chinesischen Regierung genehmigt wurde .
Das Gen p53 wurde 2005 entdeckt, um den Stoffwechsel zu regulieren. Um von der G1-auf die S-Phase zu übertragen, müssen Zellen über eine ausreichende Rohstoffunterstützung für DNA, Organellen und Proteinsynthese verfügen. Um diesen Prozess zu regulieren, sind einige Kontrollpunkte erforderlich. Einer von ihnen ist der glukoseabhängige Kontrollpunkt bei G1 / S. Er wird durch die AMP-aktivierte Proteinkinase (AMPK) reguliert. Wenn Glukose erschöpft ist, kann AMPK das Protein p53 phosphorylieren, was wiederum einen Zellstillstand induziert und den Zelltod vermeidet., Zellen, die auf den p53-abhängigen Stillstand stoßen, treten wieder in den Zellzyklus ein, wenn Glukose ausreichend ist .
Es ist bekannt, dass die Inaktivierung von p53 für die Bildung von Tumoren notwendig ist. Bykov et al. VJ und Snydel EL et al. weisen Sie darauf hin, dass eine unsachgemäße Funktion von p53 zur Proliferation eines bestehenden Tumors führen kann . Ventura und seine Mitarbeiter haben einige Experimente durchgeführt, um diese Hypothese zu testen. Sie stellten die Funktion von endogenem p53 bei primären autochthonen Tumoren wieder her, um die Folge der p53-Reaktivierung zu untersuchen., Das Ergebnis zeigte, dass die p53-Reaktivierung für die Regression autochthoner Tumoren verantwortlich war. Das bedeutet, dass inaktiviertes p53-Protein zur Tumorentwicklung führen kann . Xue und andere Wissenschaftler führten auch ein Experiment durch, um die Konsequenz der Reaktivierung von p53 an Tumoren zu testen. Sie verwendeten reversible RNA-Interferenz (RNAi), um die Expression von endogenem p53 bei Mäusen mit Leberkrebs zu regulieren. Im Experiment wird Doxycyclin (Dox) verwendet, um p53 zu reaktivieren, da der Ausdruck von p53 vollständig unterdrückt wird, wenn Dox fehlt und schnell wiederhergestellt wird, wenn Dox hinzugefügt wird., Bei Behandlung mit Dox wurde p53 miRNA abgeschaltet, was wiederum zu einer erhöhten Expression von p53 führt. Das Ergebnis zeigte, dass die Tumoren in Dox-behandelten Mäusen nach 12 Tagen nicht mehr nachweisbar werden, während Tumoren in unbehandelten Mäusen schnell wuchsen. Um die Folge einer vorübergehenden Reaktivierung von p53 zu testen, behandelten sie Mäuse 4 Tage lang mit Dox und stoppten dann. Das Ergebnis zeigte, dass selbst eine zweitägige Behandlung zu einer Regression von Tumoren führen kann und 4 Behandlungstage zu einer vollständigen Regression der Tumore führen können., Sie wiesen auch darauf hin, dass während der Tumorregression transient reaktiviertes p53 zelluläre Seneszenz auslösen kann, nicht Apoptose. Im selben Jahr fand Hu heraus, dass die embryonale Implantation bei p53-/- weiblichen Mäusen durch den Leukämie-inhibitorischen Faktor (LIF) reguliert wird. Das LIF ist ein sekretiertes Zytokin und wichtig für die Blastozystenimplantation. Das Gen, das LIF kodiert, wird als p53-Zielgen identifiziert und die p53-Bindungsstelle befindet sich in Intron 1 sowohl bei Menschen als auch bei Mäusen .