April 13, 2017, von NCI-Mitarbeitern
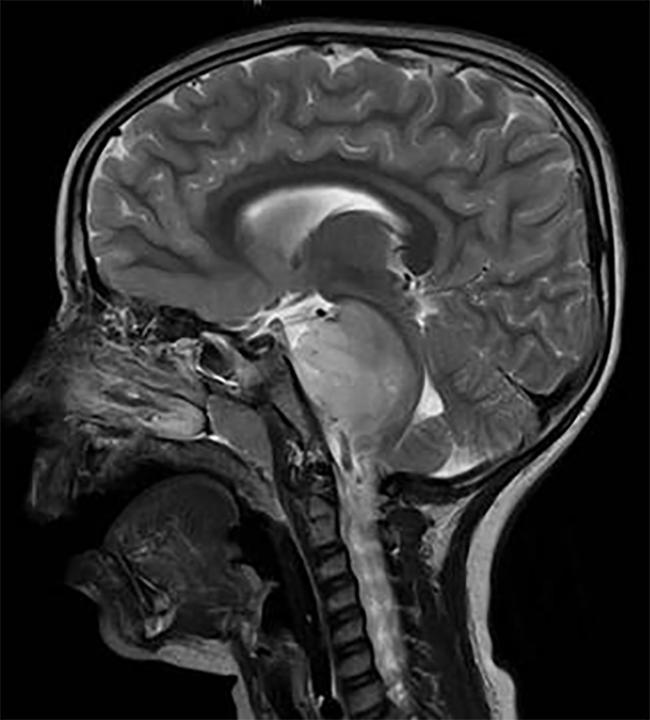
Gehirnscan eines Kindes mit einem DIPG-Tumor im Hirnstamm.
Zwei separate Studien haben festgestellt, potenzielle therapeutische Ziele in einem inoperablen pediatric brain tumor, diffuse intrinsic pontine glioma (DIPG). Das Blockieren dieser Ziele mit Untersuchungsmedikamenten verlangsamte das Tumorwachstum in Tiermodellen von DIPG.
DIPG, ein Tumor, der sich im Hirnstamm befindet, ist fast gleichmäßig tödlich.,
„Als pädiatrischer Neuroonkologe ist es einer der verheerendsten Tumoren, die wir sehen“, sagte Pratiti Bandopadhayay, M. B. B. S., Ph. D. des Dana-Farber/Boston Children ‚ s Cancer and Blood Disorders Center, der nicht an den Studien beteiligt war. „Wir haben überhaupt keine heilenden Behandlungen für diese Tumoren.“
Während die Mehrzahl der DIPG-Tumoren eine spezifische genetische Mutation trägt, war bisher unklar, welche Rolle das mutierte Protein bei der Tumorentwicklung spielt und ob seine Funktion durch Therapien gezielt werden könnte.,
Für beide neuen Studien untersuchten die Forscherteams die Biologie menschlicher DIPG-Zellen mit dieser Mutation und identifizierten Merkmale, die sie anfällig für eine Behandlung machen können. Verglichen mit Kontrollbehandlungen, Sie fanden heraus, Medikamente, die als PRC2-und BET-Hemmer bekannt sind, verringerten DIPG-Tumoren in Mausmodellen und verlängerten das Leben der Mäuse.
Die Ergebnisse beider Studien, eine von einer Forschungsgruppe an der Northwestern University und eine von einer Gruppe an der Universität Kopenhagen in Dänemark, wurden im Februar 27 in Nature Medicine veröffentlicht.,
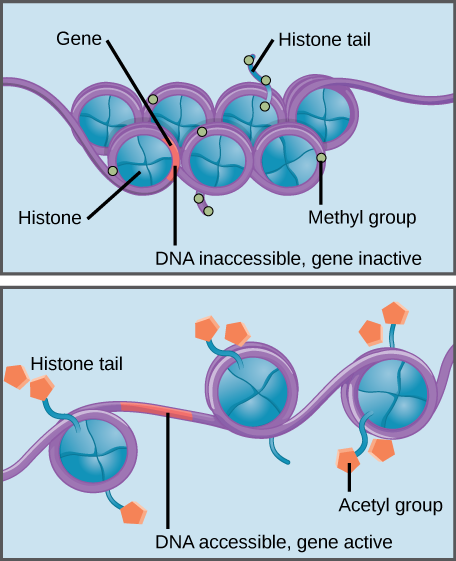
Die Histonmethylierung verstärkt die Histon–DNA-Interaktion und macht assoziierte Gene für die Genexpression unzugänglich (inaktiv). Die Histonacetylierung schwächt die Wechselwirkung und macht assoziierte Gene für die Genexpression zugänglich (aktiv).
Identifizieren eines Ziels
Vor etwa 5 Jahren arbeiteten Forscher an der St., Das Jude-Washington University Pediatric Cancer Genome Project entdeckte, dass fast 80% der DIPG-Tumoren eine spezifische Mutation im Gen für ein Protein namens Histon H3 aufweisen.
Dass ein so hoher Prozentsatz von Tumoren dieselbe Mutation aufweist, überraschte die DIPG-Forschungsgemeinschaft, sagte Ali Shilatifard, Ph. D., von der Northwestern University Feinberg School of Medicine, leitender Ermittler für eine der Studien.
Histone sind eine Familie von Proteinen, die helfen, DNA in kompakte Strukturen zu verpacken., Kurze DNA-Abschnitte wickeln sich wie Fäden auf einer Spule um Histonproteine, und Tausende von DNA-gewickelten Histonen (Nukleosomen genannt) bilden jedes Chromosom.
Spezifische Modifikationen an Histonen können die Genexpression fördern oder verhindern. Zum Beispiel schwächt die Bindung chemischer Verbindungen, die Acetylgruppen an Histone genannt werden, ihre Wechselwirkung mit DNA und fördert die Genexpression. Andererseits führt die Zugabe von Methylgruppen zu Histonen normalerweise dazu, dass sich DNA enger um Histone wickelt und die Genexpression verhindert.,
Die Identifizierung des Vorhandenseins einer Histon-H3-Mutation bei Patienten mit DIPG war der erste Schritt, sagte Dr. Bandopadhayay. „Aber um Tumore mit dieser Veränderung behandeln zu können, muss man verstehen, was die Mutation tut“, fügte sie hinzu.
In einer früheren Studie verwendeten Dr. Shilatifard und seine Kollegen Fruchtfliegen, um die Funktion des mutierten Histon-H3-Gens zu untersuchen. Im Vergleich zu Fliegen mit normalen Histon-H3-Proteinen hatten Fliegen mit der mutierten Version mehr Histone, die mit Acetylgruppen (acetyliert) besetzt waren., Diese acetylierten Histone wurden wiederum durch Moleküle gebunden, die als Bromodomain-haltige (BRD) Proteine bezeichnet werden und die Genexpression regulieren.
Für ihre aktuelle Studie, die teilweise von NCI finanziert wurde, wollten die Nordwestforscher herausfinden, ob das mutierte Histon in menschlichen Zellen die gleiche Funktion hatte. In der Tat fanden sie heraus, dass menschliche Darmkrebs-oder Nierenkrebszellen, die zur Expression des mutierten Histon-H3-Gens verändert wurden, mehr acetylierte Histone aufwiesen als Zellen, die das normale Histon-H3-Gen exprimierten.
Der erste Autor, Andrea Piunti, Ph. D.,, untersuchte dann alle mutierten H3-Histone, die sich in den Genomen von DIPG-Tumorzellen von drei Patienten befinden. Seine Analyse ergab, dass viele Nukleosomen, die mutierte Histone enthielten, acetyliert und durch BRD-Proteine gebunden waren.
Zusätzliche Experimente zeigten, dass BRD-Proteine eine direkte Rolle beim DIPG-Tumorwachstum spielen können. Die Behandlung von menschlichen DIPG-Zellen mit Untersuchungsmedikamenten, die BRD-Proteine blockieren, sogenannte BET-Inhibitoren, verlangsamte das DIPG-Zellwachstum im Vergleich zu einer Kontrollbehandlung., Und in Studien an Mäusen mit menschlichen DIPG-Zellen, die in ihre Hirnstammzellen implantiert wurden, hatten diejenigen, die mit BET-Hemmern behandelt wurden, kleinere Tumoren und lebten länger als Mäuse, die mit der Kontrollbehandlung behandelt wurden.
Diese Experimente „legen nahe, dass BET-Inhibitoren einen potenziellen therapeutischen Ansatz für DIPG bieten“, sagte Dr. Shilatifard. Präklinische Studien haben gezeigt, dass verschiedene Krebsarten—einschließlich Leukämie und Glioblastom—auch empfindlich auf BET-Inhibitoren reagieren, fügte er hinzu.,
Ein zusätzliches Ziel
Die Analyse der Northwestern Group von humanen DIPG-Tumorzellen mit mutiertem Histon H3 ergab auch, dass neben mehr Histon H3 Acetylierung viele Histone mit Methylgruppen (methyliert) verziert waren. Darüber hinaus fanden sie heraus, dass ein Enzym namens PRC2—das Methylgruppen an Histon—H3-Proteine bindet-in der Nähe vieler dieser Histone vorhanden war.
Diese Ergebnisse veranlassten die nordwestlichen Forscher, sich weiter mit der PRC2-Aktivität zu befassen., Sie fanden heraus, dass die Blockierung der PRC2—Aktivität—entweder genetisch oder mit einem PRC2-Inhibitor namens Tazemetostat-das DIPG-Zellwachstum reduzierte.
„Unerwartet zeigen diese Ergebnisse eine Rolle für die PRC2-Funktion bei der Aufrechterhaltung des DIPG-Wachstums“, schrieb die Northwestern Group.
Der Kopenhagen-Gruppe fand auch Belege für PRC2″s Rolle bei der Förderung der DIPG tumor-Wachstum. Sie beobachteten, dass Tazemetostat und ein anderer PRC2-Inhibitor das Wachstum von Maus-Gehirnzellen oder menschlichen DIPG-Zellen mit mutierten Histonen reduzierten, während die Kontrollbehandlung dies nicht tat.,
Und wenn sie Mausgehirnzellen mit mutierten Histonen in das Gehirn normaler Mäuse oder Mäuse implantierten, in denen PRC2 genetisch blockiert war, lebten diejenigen, denen die PRC2-Aktivität fehlte, länger.
Im Gegensatz dazu ergab eine separate Studie einer Forschungsgruppe in Deutschland, dass Zellen verschiedener Patienten mit DIPG gegenüber Tazemetostat nicht empfindlich waren, obwohl ihre Tumoren die Histon-H3-Mutation aufwiesen.
Ein Schritt nach vorne
Jahrzehntelange klinische Studien haben gezeigt, dass DIPG-Tumoren gegenüber einer traditionellen Chemotherapie unempfindlich sind, erklärte Dr. Bandopadhayay., Und obwohl Strahlung Symptome lindern kann, ist sie nicht heilend, und die meisten Kinder mit DIPG sterben innerhalb von 2 Jahren nach der Diagnose.
Im Jahr 2015 hat sich ein internationales Konsortium von DIPG-Forschern zusammengeschlossen, um alle verfügbaren DIPG-Zellproben zu untersuchen. Ihre Studie ergab, dass DIPG-Zellen, die von mehreren verschiedenen Patienten gesammelt wurden, durch ein histonmodifizierendes Medikament namens Panobinostat (Farydak®) abgetötet wurden. Panobinostat blockiert Enzyme, die Acetylgruppen zu Histonen hinzufügen., Jetzt testen Forscher, die Teil des NCI-unterstützten Pediatric Brain Tumor Consortium sind, die Sicherheit und beste Dosis von Panobinostat für Kinder mit DIPG in einer klinischen Phase-I-Studie.
Die Ergebnisse beider Studien deuten nun auf potenziell vielversprechende neue Behandlungsstrategien hin, sagte Dr. Shilatifard.
“ Wir wollen Betainhibitoren in eine Phase-I-Studie zur Behandlung von DIPG überführen, und unsere Kollegen am Ann & Robert H. Lurie Children ’s Hospital of Chicago treiben diese Studien voran“, sagte er.,
Darüber hinaus testen mehrere laufende klinische Studien Tazemetostat, den PRC2-Inhibitor, sowohl bei erwachsenen als auch bei pädiatrischen Patienten mit verschiedenen Krebsarten. In keiner aktuellen Studie wird Tazemetostat jedoch speziell bei Kindern mit DIPG untersucht.
Es gibt immer noch viele unbeantwortete Fragen, sagte Dr. Bandopadhayay, einschließlich, ob diese Untersuchungsmedikamente die Fähigkeit haben, die Blut–Hirn-Schranke zu überqueren und menschliche Hirntumoren zu erreichen.
Dr., Bandopadhayayay und ihre Kollegen lernen auch mehr über die DIPG-Biologie, indem sie Tumorzellen untersuchen, die aus Biopsien gewonnen wurden, zusätzlich zu denen, die aus Autopsien gewonnen wurden. Die Gewinnung von Biopsieproben wurde erst kürzlich durch wesentliche Verbesserungen der neurochirurgischen Techniken ermöglicht, erklärte sie.
„Es ist eine aufregende Zeit“, sagte sie. „Es ist gerade viel Arbeit im Gange. Das ultimative Ziel ist es, diese Kinder so behandeln zu können, dass sie eine Heilungschance mit minimalen Nebenwirkungen haben.“