Una procedura automatizzata scritta in Microsoft Visual Basic (VB) 6.,0 aggiorna DAVID settimanalmente con le seguenti procedure: chiamare una serie di applicazioni Perl e Java che scaricano dati pubblici tramite protocolli di trasferimento file anonimi (FTP) (Tabella 1); decomprimere e analizzare i dati di annotazione desiderati; creare file di dati delimitati da tabulazioni pronti per l’importazione di database; e importare i dati in un sistema di gestione di database relazionale Oracle 8i (RDBMS) Il server Web IIE di Microsoft e la tecnologia Active Server Page vengono utilizzati per accedere al database utilizzando JavaBeans e SQL (structured Query Language)., I numeri LocusLink per i set di sonde Affymetrix derivano dalle associazioni dell’Università del Michigan o NetAffx . Le annotazioni funzionali e i riferimenti incrociati del database derivano da LocusLink, che fornisce rappresentazioni stabili e curate dall’uomo dei geni. Per informazioni più dettagliate sulle fonti di dati utilizzate da DAVID si prega di consultare la sezione FAQ a .,
moduli di Analisi
DAVID si compone di quattro moduli principali: Strumento di Annotazione, GoCharts, KeggCharts, e DomainCharts. Lo strumento di annotazione è un metodo automatizzato per l’annotazione funzionale delle liste di geni. Qualsiasi combinazione di dati di annotazione può essere scelta tra 10 opzioni selezionando le caselle di controllo appropriate (Tabella 2)., Le annotazioni vengono aggiunti alla lista gene inviato selezionando il pulsante carica, che restituisce una tabella HTML contenente l”elenco originale dell” utente di identificatori aggiunto con le annotazioni funzionali scelti. I geni non annotati sono inclusi nell’output senza dati aggiunti per scopi di tracciamento.,
Il GoCharts modulo consente di visualizzare graficamente la distribuzione dei geni differenzialmente espressi tra categorie funzionali utilizzando il vocabolario controllato di Gene Ontology Consorzio (GO), che fornisce un linguaggio strutturato che può essere applicato alle funzioni dei geni e delle proteine in tutti gli organismi, anche come conoscenza continua ad accumulare e di cambiamento ., Il linguaggio è strutturato in un grafico aciclico diretto (DAG), in cui la specificità del termine aumenta e la copertura del genoma diminuisce man mano che si scende nella gerarchia. In contrasto con una vera gerarchia, i termini figlio in un DAG possono avere più di un termine genitore e possono avere una diversa classe di relazione con i suoi diversi genitori. La struttura di GO inizia con tre categorie principali, processo biologico, funzione molecolare e componente cellulare., Il processo biologico comprende gli ampi obiettivi biologici, quali la mitosi o il metabolismo delle purine, che sono compiuti dalle assemblee ordinate delle funzioni molecolari. La funzione molecolare descrive i compiti eseguiti dai singoli prodotti genici; esempi sono il fattore di trascrizione e l’elicasi del DNA. Il tipo di classificazione dei componenti cellulari comprende strutture subcellulari, posizioni e complessi macromolecolari; esempi includono nucleo, telomeri e complesso di riconoscimento dell’origine., Dopo aver scelto un tipo di classificazione, i livelli che determinano la copertura e la specificità dell’elenco vengono scelti selezionando il pulsante di opzione appropriato. Livello 1 fornisce la più alta copertura lista con la minor quantità di specificità termine. Ad ogni livello crescente la copertura diminuisce mentre la specificità aumenta in modo che il livello 5 fornisca la minor quantità di copertura con la più alta specificità a termine.
I dati di classificazione vengono visualizzati come un grafico a barre, in cui la lunghezza della barra rappresenta il numero di identificatori genetici in ciascuna categoria., L’utente può impostare parametri di visualizzazione per l’ordinamento dei dati di output e la visualizzazione di categorie che contengono almeno un numero minimo di geni. Selezionando una singola barra si apre una nuova tabella HTML che visualizza l’identificatore del gene, il numero LocusLink, il nome del gene, la classificazione corrente e altre classificazioni per ciascun gene in quella categoria. Un pulsante “Mostra tutto” apre una nuova tabella HTML che visualizza tutti i dati di classificazione e un pulsante “Mostra dati grafico” apre una tabella HTML contenente i dati del grafico sottostanti, consentendo così agli utenti di ricreare grafici personalizzati in un foglio di calcolo., Un nuovo grafico può essere visualizzato per qualsiasi sottoinsieme di geni selezionando il tipo di classificazione e il livello utilizzando le caselle di controllo e pulsanti di opzione disponibili all”interno della pagina corrente dell” utente che consentono funzionalità di drill-down. Un conteggio del numero di geni annotati è incluso nell’output e i geni non annotati sono raggruppati nella categoria “non classificati”, fornendo così agli utenti un sistema di tracciamento automatico per i geni non annotati.
KeggCharts mostra graficamente la distribuzione dei geni espressi in modo differenziale tra i percorsi biochimici di KEGG., Ogni percorso è collegato alla mappa del percorso di KEGG, in cui i geni espressi in modo differenziato dalla lista originale sono evidenziati in rosso. In questa vista i geni sono ulteriormente collegati a annotazioni aggiuntive disponibili attraverso il sistema di recupero DBGET di KEGG . Come con GoCharts, l’utente può impostare i parametri di visualizzazione per l’ordinamento dei dati di output e la visualizzazione di categorie che contengono almeno un numero minimo di geni e la visualizzazione KeggCharts eredita tutte le caratteristiche dinamiche di GoCharts.
DomainCharts mostra la distribuzione di geni espressi in modo differenziato tra i domini proteici PFAM ., Ogni denominazione di dominio è collegata al Conserved Domain Database (CDD) del National Center for Biotechnology Information (NCBI), dove i dettagli riguardanti la funzione del dominio, la struttura e la sequenza sono prontamente disponibili. Come con GoCharts e KeggCharts, l’utente può impostare i parametri di visualizzazione per l’ordinamento dei dati di output e la visualizzazione di categorie che contengono almeno un numero minimo di geni e la DomainCharts visualizzazione eredita tutte le caratteristiche dinamiche di GoCharts e KeggCharts. Per ulteriori informazioni sulla funzionalità di DAVID visita la sezione FAQ all’indirizzo .,
Utilizzando DAVID per estrarre l’annotazione funzionale
Per dimostrare la funzionalità di DAVID abbiamo analizzato un elenco di geni espressi in modo differenziale nelle cellule mononucleate del sangue periferico umano (PBMC) dopo l’incubazione con le proteine dell’involucro dell’HIV-1. I dettagli delle procedure sperimentali, di preparazione dell’RNA e di ibridazione GeneChip, insieme ai dettagli delle normalizzazioni chip-to-chip e all’analisi statistica dell’espressione genica differenziale sono forniti in Cicala et al. ., In breve, le PBMC umane primarie e i macrofagi derivati dai monociti sono stati incubati per 16 ore con la proteina inviluppo HIV-1 (gp120). Microarray oligonucleotidici ad alta densità (Affymetrix HU-95A GeneChip) sono stati utilizzati per monitorare gli eventi trascrizionali indotti da gp120. Questa analisi ha portato all’identificazione di 402 geni espressi in modo differenziale.
Mentre 16 geni modulati dall’HIV-1 gp120 sono stati precedentemente associati alla replicazione dell’HIV e / o alla segnalazione di inviluppo, i geni rimanenti hanno una funzione sconosciuta o non sono mai stati associati all’HIV-1 o al gp120., La conversione di questo elenco di geni in significato biologico richiede la raccolta di informazioni pertinenti da diversi archivi di dati. Per molti ricercatori questo processo consiste nella navigazione iterativa attraverso diversi database per ciascun gene, raccogliendo manualmente informazioni specifiche sul gene per quanto riguarda la sequenza, la funzione, il percorso e l’associazione della malattia. Al contrario, l’approccio sistematico di DAVID aggiunge simultaneamente informazioni biologicamente ricche derivate da diverse fonti di dati pubblici agli elenchi di geni in parallelo., Selezionando lo strumento di annotazione di DAVID e caricando l “elenco di 402 geni differenzialmente espressi avvia l” annotazione funzionale e l “analisi dell” intero set di dati. Una volta inviato, l’elenco dei geni viene memorizzato per l’intera sessione di analisi, consentendo agli utenti di passare da un modulo all’altro senza dover inviare nuovamente i dati.
Strumento di annotazione
Lo strumento di annotazione fornisce diverse opzioni di annotazione e crea una vista tabulare dell’elenco dei geni degli utenti e delle annotazioni disponibili (Tabella 2)., La scelta dei campi di annotazione Gene Symbol, LocusLink, OMIM, Unigene, Reference Sequence e Gene Name seguita selezionando il pulsante” Upload ” produce una tabella HTML nel browser web contenente tutti i geni e le loro annotazioni disponibili, dove gli identificatori dei geni, i dati descrittivi e di classificazione vengono estratti dal database e aggiunti all’elenco dei geni (Figura 1). Gli identificatori del gene quali il simbolo del gene ed il LocusLink hyperlinked ai dati gene-specifici supplementari disponibili alle loro fonti originali, così fornendo i dettagli gene-specifici approfonditi ed i pedigree dell’annotazione., Dati di classificazione e riassunti funzionali possono essere utilizzati per eseguire rapidamente la scansione di informazioni rilevanti per il sistema sperimentale del ricercatore. Il tempo del server richiesto per l’esecuzione di questo modulo è correlato linearmente con la dimensione dell’elenco dei geni e richiede meno di 45 secondi per elenchi fino a 1.000 geni (Figura 2, i numeri tra parentesi rappresentano i valori r2). Questi risultati dimostrano la potenza e l’efficienza di un approccio integrato all’annotazione funzionale di grandi set di dati.,

Output dello strumento di annotazione. Vengono mostrate le annotazioni aggiunte per i primi set di probe Affymetrix in una tabella HTML contenente tutte le 402 voci. Le informazioni categoriali sulle condizioni sperimentali sono state inviate insieme agli identificatori Affymetrix probe-set e incluse nell’output nella colonna valore. Gli identificatori come Symbol, LocusLink, OMIM, RefSeq e Unigene accession sono iper-collegati alle loro origini di origine per informazioni più dettagliate., Testo incluso nei campi di riepilogo è derivato da descrittivo, informazioni funzionali fornite nei rapporti LocusLink di NCBI.
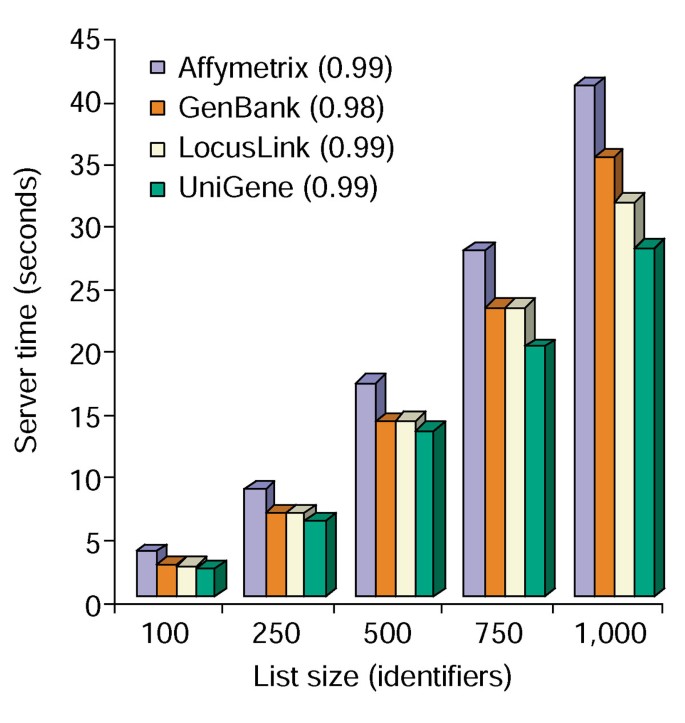
l’analisi di Strumento di Annotazione. Tempo del server richiesto (asse y) per aggiungere simultaneamente tutte le 10 opzioni di annotazione agli elenchi di geni di dimensioni comprese tra 100 e 1.000 (asse x)., La media di tre studi per liste di geni contenenti Affymetrix, GenBank, LocusLink, e unigene identificatori sono mostrati e i numeri tra parentesi rappresentano il valore r2 della correlazione tra dimensione gene-list e il tempo del server richiesto per l’annotazione.
GoCharts
Scegliendo il modulo GoCharts si apre una nuova finestra con una varietà di opzioni., Gli utenti scelgono tra tre tipi generali di classificazione (processo biologico, funzione molecolare e componente cellulare) e cinque livelli di annotazione che rappresentano la copertura del termine e la specificità (vedi sezione Moduli di analisi). È possibile specificare qualsiasi combinazione di classificazione e livello di copertura. Sono incluse anche le opzioni per annotare elenchi di geni con tutti i termini GO disponibili o solo i termini più specifici, che sono indicati come nodi terminali., L’opzione di scegliere diversi livelli di specificità del termine fornisce la flessibilità necessaria e consente quindi ai ricercatori di determinare dinamicamente quale livello di copertura e specificità si adatta meglio ai loro dati e allo stadio di analisi. Ad esempio, le analisi in fase iniziale possono consistere nell’annotare elenchi di geni con termini molto generali al fine di ottenere un’ampia comprensione dei dati. In questo caso, selezionando il processo biologico e il livello 1 classifica i geni usando termini generali come” morte “e”comunicazione cellulare”., L’utilizzo di una maggiore specificità del termine facilita l’estrazione di informazioni funzionali più dettagliate. In questo caso la selezione del processo biologico e il livello 5 classificano i geni usando termini come “cambiamenti mitocondriali apoptotici” e “percezione chemiosensoriale”.
Tuttavia, l’aumento della specificità del termine ha un costo, in quanto aumenta la copertura della lista diminuisce (Figura 3). Nei nostri studi troviamo che il livello 2 in genere mantiene una buona copertura fornendo anche una significativa specificità a termine., La figura 4a illustra come la visualizzazione di GoCharts rivela rapidamente che 35 geni espressi in modo differenziato sono coinvolti nelle”risposte allo stress”. Ogni termine GO può essere visualizzato nell’albero o nelle viste DAG tramite collegamenti ipertestuali a QuickGO .
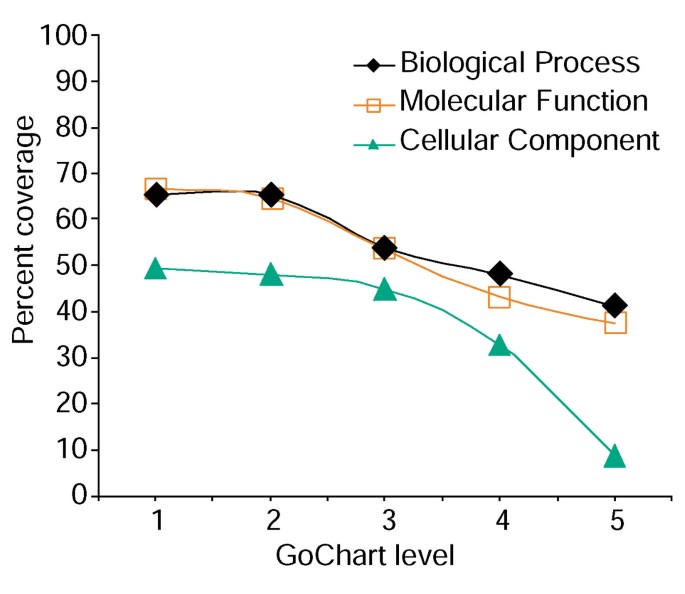
Analisi della copertura della lista dei geni utilizzando GoCharts. Un elenco di 402 identificatori del set di probe Affymetrix è stato annotato con le classificazioni funzionali assegnate al Proteoma fornite da LocusLink., La copertura percentuale rappresenta il numero di geni su 402 che sono stati annotati a un livello di specificità del termine all’interno del processo biologico, della funzione molecolare e dei tipi di classificazione dei componenti cellulari. La copertura percentuale diminuisce all’aumentare della specificità del termine.
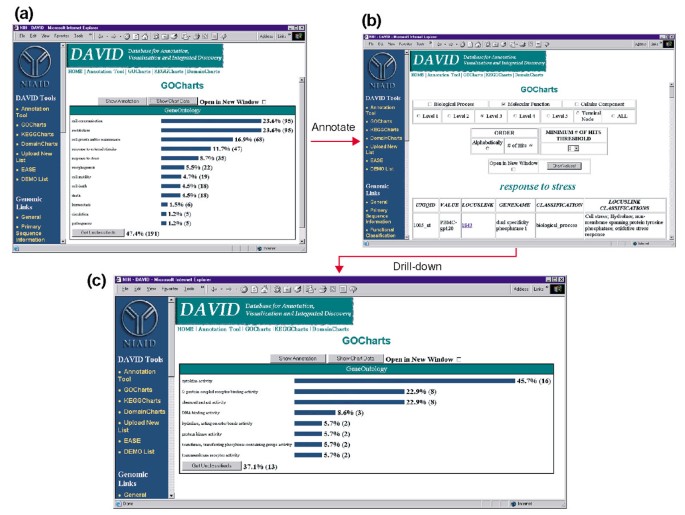
Uscita di GoCharts. (a) Un grafico a barre che mostra la distribuzione di geni espressi in modo differenziale tra i processi biologici dell’ontologia genica (GO)., I parametri sono stati impostati per PASSARE al livello 2, una soglia di hit di cinque e l’output è stato ordinato per numero di hit. Le barre blu sono collegate a dati di annotazione aggiuntivi mostrati in (b). Selezionando la barra blu in (a) corrispondente a “risposta allo stress” si apre una tabella HTML che mostra il LocusLink, il nome del gene, la classificazione corrente e altri dati di classificazione per i geni in quella categoria. (c) Questo sottoinsieme di geni coinvolti nella “risposta allo stress” è stato ulteriormente caratterizzato selezionando la funzione molecolare GO, il livello GO 3, una soglia di hit di 2 e ordinati per numero di hit., Selezionando il pulsante “Valori grafico” si crea un nuovo istogramma che rivela che 16 dei 35 geni di risposta allo stress codificano proteine che possiedono attività citochinica.,
a Causa di HIV-1 ha un impatto importante sulla funzione delle cellule del sistema immunitario e la loro capacità di svolgere le risposte di stress, abbiamo selezionato la barra dell’istogramma che rappresenta il numero di geni coinvolti nella risposta allo stress, che apre una tabella HTML che contiene il Affymetrix identificatore, LocusLink numero, il nome del gene, l’attuale classificazione, e altre classificazioni per tutti i 35 geni (Figura 4b)., Ora che abbiamo ridotto la nostra lista di geni a quei geni coinvolti nelle risposte allo stress, abbiamo ulteriormente caratterizzato questo sottoinsieme ripetendo la procedura GoCharts disponibile nella parte superiore della tabella HTML stress-response. Scegliendo la funzione molecolare, il livello 3 produce un nuovo istogramma che rivela rapidamente che quasi la metà (16/35) dei geni di risposta allo stress possiede attività citochinica (Figura 4c)., In effetti, le citochine hanno dimostrato di svolgere un ruolo importante nel ciclo di vita dell’HIV-1 e i risultati ottenuti qui suggeriscono che il trattamento delle PBMC con proteine dell’involucro dell’HIV-1 modula significativamente la trascrizione di numerosi geni di citochine. L’efficienza con cui GoCharts ha sistematicamente riassunto questo grande set di dati con visualizzazioni grafiche, pur rimanendo legato ai dati primari e alle risorse esterne ha migliorato drasticamente il processo di discovery.,
KeggCharts
La figura 5a descrive l’output di KeggCharts con un istogramma che mostra la distribuzione dei geni espressi in modo differenziale tra i percorsi biochimici. Il grafico mostra che una via di KEGG di apoptosi include cinque geni indotti da HIV-1 gp120. Selezionando il nome del percorso si apre la corrispondente mappa del percorso biochimico di KEGG e si evidenziano in rosso i geni espressi in modo differenziale che funzionano in quel percorso (Figura 5b). In questa vista i geni sono ulteriormente collegati a annotazioni aggiuntive disponibili attraverso il sistema di recupero DBGET di KEGG ., Si noti che solo quattro geni nella via dell’apoptosi di KEGG sono evidenziati in rosso, mentre lo strumento KeggCharts ha mappato cinque set di sonda Affymetrix sulla via dell’apoptosi. Questa differenza è dovuta al fatto che due dei probeset Affymetrix stanno prendendo di mira lo stesso gene “TNF-alfa”.
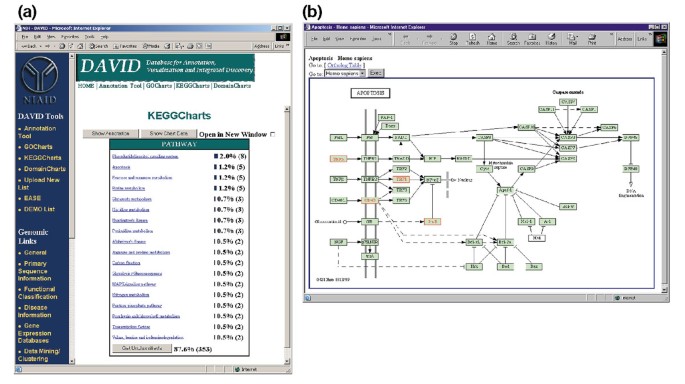
Output di KeggCharts. (a) Grafico di visualizzazione che mostra la distribuzione di 402 geni tra le vie biochimiche di KEGG. La soglia hit è stata impostata su tre e l’output è stato ordinato per numero di hit., Il gran numero di identificatori non classificati è dovuto al fatto che KEGG è centrica della via biochimica e quindi fornisce una bassa copertura delle liste di geni. Analogamente all’output di GoCharts, le barre blu rappresentano il numero di geni in ciascun percorso. Selezionando una barra blu si apre una tabella HTML che mostra il LocusLink, il nome del gene, la classificazione corrente e altri dati di classificazione per i geni in quel percorso (dati non mostrati)., (b) La via biochimica di KEGG che appare dopo la selezione del nome della via “apoptosi” in (a) raffigura quattro geni espressi in modo differenziato all’interno della via apoptosi evidenziandoli in verde chiaro e rosso. Il fatto che il percorso di KEGG evidenzia solo quattro geni mentre il KeggChart mappa cinque set di sonda Affymetrix al percorso di apoptosi è dovuto al fatto che due set di sonda mirano allo stesso gene “TNF-alfa”.,
DomainCharts
I DomainCharts sono operativamente simili sia ai KeggCharts che ai GoCharts, tranne per il fatto che i risultati descrivono visivamente la distribuzione dei geni tra i domini proteici PFAM (Figura 6a). L’istogramma DomainCharts identifica 16 geni con domini chinasi (pkinasi), probabilmente riflettendo gli effetti dell’HIV-1 gp120 sul macchinario di trasduzione del segnale. Il grafico identifica anche sei geni con domini di interleuchina-8 (IL-8), un dominio che rappresenta un motivo altamente conservato tra le citochine di risposta allo stress., Selezionando il nome di dominio ” IL8 ” si apre la pagina Conserved Domain Database (CDD) corrispondente a quel dominio PFAM (Figura 6b). Questa pagina fornisce sequenza dettagliata, struttura e informazioni funzionali sul dominio IL-8 e le proteine che lo contengono.
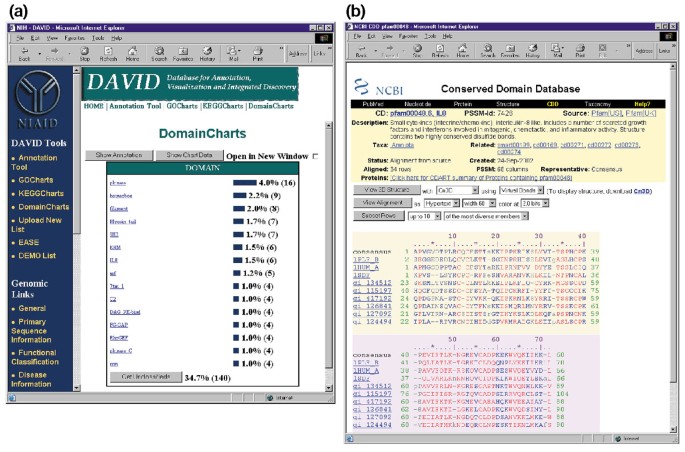
Output di DomainCharts. (a) Grafico di visualizzazione che mostra la distribuzione di 402 geni tra i domini proteici. I parametri sono stati impostati su una soglia minima di hit di quattro e l’output è stato ordinato per numero di hit., Simile all’output di GoCharts e KeggCharts, le barre blu rappresentano il numero di geni contenenti quel particolare dominio. Selezionando una barra blu si apre una tabella HTML che mostra il LocusLink, il nome del gene, la classificazione corrente e altri dati di classificazione per i geni in quel percorso (dati non mostrati)., (b) la Selezione del nome di dominio “IL8” in (a), che contiene sei geni differenzialmente espressi, porta l’utente ad una nuova pagina contenente l’uscita dal Dominio Conservato Database (CDD) di NCBI, che fornisce informazioni dettagliate circa l’il-8 di dominio, incluse le informazioni strutturali, di allineamenti multipli di sequenza, e descrittivo informazioni sul dominio e le proteine che possiedono.