Il gene p53 è stato scoperto per la prima volta nel 1979. Una proteina è stata identificata nelle cellule di topo trasformate dal virus simian 40 (SV40) mediante immunoprecipitazione con siero anti-T; questa proteina è stata chiamata proteina p53 . Nello stesso anno, Kress e altri scienziati hanno trovato una nuova classe di proteine con una massa molecolare che va da 50-60kDa. Questo tipo di proteina è stata poi identificata come p53 . La proteina p53 può anche essere identificata da varie linee cellulari trasformate mediante immunoprecipitazione., Lane e Linzer hanno anche ottenuto un risultato simile nel 1979. Altre prove per identificare p53 è che p53 è stato espresso in tutte le cellule di topo trasformate testate; questi test includono sarcomi indotti chimicamente, fibroblasti trasformati e leucemie, mentre nelle cellule normali, p53 non è stato espresso. Inoltre, un alto livello di p53 è stato rilevato nella maggior parte delle cellule trasformate, indipendentemente da come le cellule sono state trasformate, spontaneamente o non spontaneamente ., Ciò era in gran parte dovuto alla maggiore stabilità del p53, tuttavia, nelle cellule carcinoa embrionali F9, esprimeva un alto livello di p53, ciò era dovuto alla quantità di mRNA p53 tradotto .
Dopo che la proteina p53 era stata scoperta nel 1979, divenne popolare analizzarla. Tuttavia, a quel tempo, poiché era una proteina appena scoperta, e non c’era un nome precedente per esso, diverse istituzioni usavano nomi diversi e pubblicavano documenti con nomi diversi., Per risolvere questo problema, nel 1983, durante il 1st International p53 workshop tenutosi a Oxted, Regno Unito, scienziati di diversi gruppi di ricerca in diversi paesi si sono riuniti per discutere una nomenclatura comune per questa proteina appena scoperta. In questo incontro,” p53 ” diventa il suo nome ed è stato usato da allora. Si è pensato che il motivo per cui gli scienziati hanno chiamato la proteina p53 è che la massa molecolare di questa proteina è 53kDa che si basa sulla sua migrazione in gel SDS. Successivamente la massa molecolare è stata dimostrata errata e la massa molecolare corretta dovrebbe essere 43.,7kDa perché p53 contiene una regione ricca di prolina, e questa regione può ridurre la migrazione di p53 in SDS gel. Ma il nome “p53” è rimasto .
Durante il 1980, la proteina p53 è stato creduto di essere coinvolto nel ciclo cellulare, oltre a svolgere un ruolo nella replicazione del DNA. Più tardi, nel 1982-1994, le persone hanno scoperto che alcune oncoproteine virali erano in grado di legarsi a p53, formando un complesso. Nel 1982, Sarnow et al. ha scoperto che l’adenovirus E1b (58kDa) può interagire con una proteina 54kDa presente nelle cellule di topo trasformate SV40 menzionate sopra., Secondo i risultati delle specificità immunologiche degli anticorpi T e delle mappe peptidiche della proteina 54kDa, questa proteina 54kDa è identificata come p53 . Nello stesso anno, gli scienziati hanno scoperto che se iniettassero l’anticorpo p53 nelle cellule di topo Swiss 3T3, inibirebbero le cellule che entrano nella fase S del ciclo cellulare, tuttavia; nella stessa situazione, l’anticorpo p53 non ha influenzato la sintesi del DNA indotta da SV40 o adenovirus .,
Più tardi nel 1984, gli scienziati hanno esaminato l’effetto del p53 sui fibroblasti 3T3 non trasformati; hanno analizzato il tasso di sintesi della proteina p53 in diversi punti temporali e hanno scoperto che nella fase G1 tardiva, il tasso di sintesi e il livello della proteina p53 e il suo mRNA correlato aumentano. Questo risultato suggerisce che la proteina p53 inibisca le cellule che entrano nella fase di divisione dall’interfase . Maltzman W et al. ha fatto un altro esperimento nello stesso anno. Hanno trattato la cellula di topo non trasformata con luce UV e cancerogeno chimico UV-mimetico 4NQO e hanno rilevato un alto livello di p53., Il risultato ha mostrato che l’elevata espressione di p53 non è solo un simbolo che indica il ciclo cellulare, ma anche, cosa più importante, un componente coinvolto nella sintesi del DNA e nella proliferazione cellulare . Nel 1987, studiando il complesso dell’antigene T del virus simian 40 e della DNA polimerasi α, Gannon e altri scienziati hanno trovato un cambiamento simile nell’antigene quando legato a p53 e polimerasi α. Hanno anche scoperto che ad una certa concentrazione dei tre componenti, possono formare uno speciale complesso trimerico che include l’antigene T, p53 e la DNA polimerasi α., Poiché l’antigene T è coinvolto nella replicazione del DNA virale e nella trasformazione cellulare, questo risultato indica che p53 svolge un ruolo nel controllo del ciclo cellulare e della replicazione del DNA .
Come l’esperimento ha mostrato sopra, p53 ha la capacità di immortalare le cellule. Nel 1984, Eliyahu D et al. trovato che p53 e il prodotto di oncogene myc condividevano alcune proprietà simili. Entrambi hanno la capacità di legarsi ad altre proteine e sono coinvolti nel ciclo cellulare, ed entrambi si accumulano nei nuclei delle cellule trasformate ., Bienz, Pennica e Oren hanno analizzato le sequenze aminoacidiche della proteina p53 e il prodotto di myc, e hanno scoperto che le due proteine mostrano somiglianze nella struttura molecolare e nella posizione di residui carichi speciali. Quindi gli scienziati hanno proposto che p53 possa agire come un oncogene. Sulla base di questa ipotesi, Eliyahu D et al. ho fatto degli esperimenti. Poiché i fibroblasti embrionali a tasso primario possono essere trasformati dal coinvolgimento sia del prodotto myc che di Ha-ras, le cellule renali primarie del ratto del bambino possono anche essere trasformate dalla cooperazione di Ha-ras e della regione precoce dell’adenovirus 1A , Eliyahu D et al., ha deciso di utilizzare questo tipo di sistema di test biologici per identificare la funzione oncogenica del p53. Hanno trattato cellule embrionali normali con p53 e hanno attivato Ha-ras. Il risultato ha mostrato che le cellule bersaglio incontrano cambiamenti morfologici e producono alti livelli di p53, Eliyahu D et al. pensato che la trasformazione dei fibroblasti embrionali da p53 e Ha-ras spiegato che il gene p53 è un oncogene ., Nel 1985, Jenkins propose che il gene p53 potesse estendere la durata della vita delle cellule, migliorare l’affettività della trasformazione riorganizzando la sua sequenza codificante che potrebbe causare la produzione di proteine stabili .
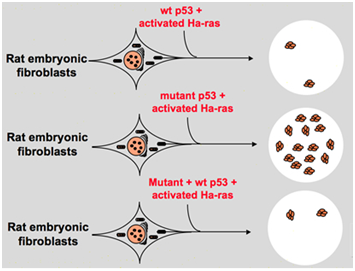
Tuttavia, alla fine degli anni 1980, gli scienziati hanno iniziato a rendersi conto che p53 è un gene soppressore del tumore invece di un oncogene. Hanno osservato che p53 con funzione normale non può essere rilevato in molti dei tumori e ha scoperto che perdere l’espressione e la funzione del gene p53 wild-type è necessario durante la trasformazione cellulare., Questi aumentano la possibilità che il gene p53 wild-type possa inibire la progressione neoplastica . Poi hanno formulato un’altra ipotesi: il gene clone p53 utilizzato in esperimenti precedenti contiene mutazioni negative dominanti all’interno del dominio altamente conservato occasionalmente, che portano a risultati dell’esperimento opposti . Nel 1988, Ben ed altri scienziati hanno individuato una quantità enorme di p53 riorganizzato in linee cellulari murine dell’eritroleukemia-DP20-1 e CB3 che sono derivati dalle spleens di murine infettate con il virus della leucemia dell’amico ., Nel 1989, Eliyahu, che ha sottolineato che p53 è un oncogene cambiato idea, e ha supposto che wild-type p53 gene può inibire la trasformazione cellulare. Eliyahu e altri scienziati hanno studiato l’effetto della proteina p53 wild-type codificata dai plasmidi e dal mutante p53 sulla capacità di suscitare la trasformazione primaria dei fibroblasti embrionali mediante varie combinazioni di oncogene in vitro. Ad esempio mutante p53 più ras e myc più ras., Il risultato ha mostrato che il p53 di tipo selvaggio porta ad un’enorme riduzione dei fuochi trasformati causati dal mutante p53 più ras; il mutante p53 non ha mostrato alcuna inibizione sui fuochi trasformati causati da myc più ras, mentre la trasformazione mediata da myc più ras è molto sensibile all’espressione di p53 di tipo selvaggio. La figura 1 mostra questo esperimento in modo conciso. Ha dimostrato che rispetto al mutante p53, wild-type p53 mostra un effetto ovviamente inibitorio sulla trasformazione cellulare. L’effetto è positivamente correlato al livello di espressione di wild-type p53 e negativamente correlato al livello di espressione del mutante p53., Questo esperimento ha suggerito che il p53 wild-type può effettivamente avere una funzione opposta rispetto al p53 mutante e può inibire la tumorigenesi . Attualmente, p53 è riconosciuto come un gene soppressore del tumore. Si stima che circa la metà dei tumori sia causata da p53. È uno dei geni mutati più frequentemente negli esseri umani e il gene analizzato più frequentemente in tutto il mondo .
Durante i primi anni del 1980, il percorso biochimico di p53 e l’effetto della mutazione p53 non erano chiari. Nel 1991, Kern e altri scienziati hanno scoperto che una sequenza di DNA a coppia di 33 basi si lega specificamente al p53 wild-type in vitro. Hanno anche scoperto che la proteina p53 contiene due mutazioni che di solito si trovano nei tumori umani che non possono legarsi a questa specifica regione del DNA. Quindi hanno supposto che la funzione di p53 dipenda dalla sua capacità di legare specifiche sequenze di DNA, e questa capacità è alterata da mutazioni trovate nei tumori umani., Suppongono anche che questa sequenza di DNA a coppia di 33 basi potrebbe non essere l’unica sequenza che ha la capacità di legarsi specificamente al p53 negli esseri umani; tuttavia, può aiutare le persone a comprendere meglio la funzione di p53 . Più successivamente, p53 è stato trovato per svolgere un ruolo durante il ciclo cellulare, la riparazione del DNA, la differenziazione, l’inizio dell’apoptosi e dell’angiogenesi. Rotter V et al. trovato che p53 up-regola la differenziazione delle cellule. Ad esempio, un alto livello di proteina p53 è stato rilevato in diversi passaggi chiave durante la differenziazione delle cellule B. Elevato p53 può anche essere rilevato durante la spermatogenesi., Nel frattempo, solo un livello molto basso di proteina p53 può essere rilevato in alcuni organi di topi adulti .
Nel 1990, uno strumento utile è stato scoperto occasionalmente. È un mutante sensibile alla temperatura di p53, chiamato p53val135. Può agire come un vero p53 wild-type alla temperatura di 32,5 oC, sopprimendo la trasformazione, e può anche agire come altro p53 mutato alla temperatura di 37,5 oC o superiore a 48oC, suscitando la trasformazione. Inoltre, per le cellule trasformate che esprimono p53val135, la sua proliferazione è controllata alla temperatura permissiva e questo tipo di controllo è reversibile., Usando questo mutante p53val135, p53 wild-type è stato scoperto per indurre l’arresto del ciclo cellulare a G1 o G2 / M . Nel 1991, Elisheva et al. ha scoperto che il p53val135 sensibile alla temperatura ha svolto una funzione diversa nella linea cellulare della leucemia mieloide murina. Dopo la riattivazione di p53val135 per alcuni giorni, tutte le cellule sono morte e questa morte mostra alcune proprietà di apoptosi . Un anno dopo, un risultato simile fu ottenuto da Shaw. Un tipo selvaggio p53 è stato trasfettato in una linea cellulare derivata dal tumore del colon umano EB., Le cellule sono state esaminate sotto luce e microscopi elettronici, e trovato per mostrare alcune proprietà di apoptosi . Nel 1990, Scheffner et al. e altri scienziati hanno scoperto che E6 che stimola la distruzione delle proteine regolatrici delle cellule ospiti è codificato dai tipi di papillomavirus umano oncogenico 16 e 18, e può formare un complesso con p53 di tipo selvaggio in vitro, che a sua volta causa la degradazione della proteina p53 .
Nel 1992, una proteina chiave MDM2 è stata scoperta perché si lega strettamente con p53 e inibisce la transattivazione mediata da p53., La massa molecolare di MDM2 è 90kDa e forma un complesso con p53 sia mutato che selvaggio . Nello stesso anno, Livingstone RL et al. ha studiato se la cellula ha perso una o entrambe le copie di alleli p53 di tipo selvaggio e se questo fosse sufficiente a causare l’amplificazione genica. L’amplificazione genica è stata rilevata principalmente nelle cellule trasformate ma non nei fibroblasti normali. Il risultato ha mostrato che le cellule che perdono una copia degli alleli p53 agiscono come p53 wild-type, mentre le cellule che perdono entrambe le copie degli alleli p53 wild type mostrano una maggiore frequenza di amplificazione . Un altro esperimento fatto da Yin Y et al., ha mostrato un risultato simile .
Nel 1993 è stato identificato un gene bersaglio p53 chiamato CDKN1A. Codifica la proteina p21 che è un inibitore della chinasi ciclina-dipendente che inibisce la ciclina-CDK2 e CDK1 legandosi a loro. Nel 1993, Szekely ha scoperto che l’antigene nucleare del virus Epstein-Barr 5 (EBNA-5) è codificato dal virus Epstein-Barr e può infettare la cellula linfoblastoide B umana. Un peptide lungo di 66 aminoacidi è responsabile della formazione di EBNA-5-p53 complesso, le mutazioni puntiformi di p53 non hanno influenzato la sua capacità di legame con EBNA-5., Tuttavia, inibisce le sue formazioni di complessi con altre molecole . Nel 1994, Cho e i suoi collaboratori descrissero per la prima volta la struttura cristallina del complesso p53-DNA. Questo dominio di legame del DNA è stato anche chiamato il dominio di base. Contiene residui 102-292 e consiste in un sandwich beta. Hanno anche dimostrato la struttura dettagliata del dominio principale . Sempre nel 1994, Wang XW et al. l’interazione tra la proteina X del virus dell’epatite B (HBX) e la proteina p53 wild-type nell’uomo., Hanno scoperto che l’HBX può inibire la capacità di p53 di legarsi ad altri DNA sequenza-specifici dopo che è legato a p53 e può anche inibire l’associazione di p53 con fattori di trascrizione .
Nel 1997, Honda R et al. in primo luogo ipotizzato che MDM2 può innescare p53 ubiquitilazione e portare alla degradazione di p53 da un sistema ubiquitina-proteasoma. Hanno sottolineato che MDM2 si lega al dominio N-terminale (NTD) di p53 e agisce come ubiquitina ligasi E3 . Sempre nel 1997, sono state scoperte due nuove famiglie di proteine, p63 e p73 che condividono una sostanziale omologia con p53., p73, chiamato anche proteina tumorale 73, è codificato da un gene situato in 1p36. La posizione viene eliminata frequentemente nel neuroblastoma e in altri tumori. p73 può attivare i geni bersaglio p53 e interagisce con p53 . Yang et al. trovato che il gene p63 si trova in 3q27-29 e può essere rilevato in varie cellule di topo e umane. Come p73, p63 può transactivate i geni dell’obiettivo p53 significativamente, può anche indurre l’apoptosi. Una caratteristica di p63 è che la maggior parte di p63 manca di un N-terminale ., Nello stesso anno, Serrano e colleghi hanno scoperto che i fibroblasti murini primari possono essere trasformati da ras in assenza di p53 o p16 e p53 o p16 inattivi possono facilitare il processo di immortalizzazione delle cellule umane. Questi risultati suggeriscono che p53 svolge un ruolo nella senescenza cellulare . Poi, nel 1997, p53 è stato trovato per svolgere un ruolo nell’inizio di apoptosi. Quando le cellule entrano nella fase di proliferazione, i telomeri alla fine di ciascun cromosoma si accorciano dopo ogni ciclo di replicazione del DNA a causa della replicazione incompleta del DNA a singolo filamento alla fine del supporto del DNA ., Il gene oncosoppressore attivato p53 limita il numero di volte in cui può verificarsi la divisione cellulare. Wynford TD ha scoperto che con la perdita della funzione di p53 wild-type, tutti i fibroblasti sfuggono all’apoptosi. Inoltre, la funzione di transattivazione di p53 può essere attivata dall’apoptosi . Wynford TD ha proposto che ci sono tre possibilità di come p53 è attivato. Il primo è la modifica post-traslazionale mediante fosforilazione, il secondo è up-regola i cofattori trascrizionali come p33ING1, l’ultimo è down-regola gli inibitori p53 come MDM2 .
Nel 2000, Brodsky MH et al., studiato gli obiettivi di trascrizione di p53 in Drosophila. Ci sono prove per mostrare che gli occhi di Drosophila mostrano un grave fenotipo dell’occhio ruvido sotto l’espressione di p53 umano che indurrà l’apoptosi delle cellule del disco visivo dell’occhio, causando la perdita di cellule pigmentate, infine inibendo lo sviluppo dell’occhio di Drosophila, quindi Drosophila può essere un animale modello per studiare la funzione di p53. Brodsky ha scoperto che il gene rpr contiene un sito di legame p53 consenso che si trova nella regione cis-regolamentazione di rpr, ed è anche un attivatore di apoptosi., Con altre prove, Brodsky ha affermato che rpr è un obiettivo trascrizionale del p53 . Nel 2001, Derry e colleghi hanno scoperto che C. elegans non ha un gene p53, ma in effetti contiene un gene cep-1 che codifica le proteine che hanno una sequenza simile con la proteina p53. Questo gene di C. elegans codifica la proteina CEP-1 che ha la capacità di indurre l’apoptosi dallo sforzo genotossico ed è una componente necessaria durante la meiosi .
Nel 2002, Tyner e colleghi hanno proposto che p53 svolge un ruolo nella regolazione dell’invecchiamento degli organismi., Per studiare la funzione di p53, hanno creato topi geneticamente modificati con p53 mutato eliminando gli esoni 1-6 e una regione a monte del gene p53 di tipo selvaggio (p53+/+), chiamato p53+/m. Agisce come p53 di tipo selvaggio e ha una maggiore resistenza ai tumori spontanei meglio di p53 di tipo selvaggio. Nell’esperimento, hanno monitorato i topi contenenti p53+ / m, p53+/+ e p53+/-. p53 + / – significa che i topi perdono una copia del gene p53 wild-type., I risultati hanno mostrato che nessuno dei topi con p53+/m ha sviluppato tumori potenzialmente letali, tuttavia, oltre l ‘ 80% dei topi con p53+/- e oltre il 45% dei topi con p53+/+ ha sviluppato questo tipo di tumori. Guardando all’interno dei tumori, sono state osservate lesioni tumorali localizzate in 2 su 35 topi p53 + / m, al contrario, vari tumori come linfomi e osteosarcomi sono stati trovati in topi p53+/- e p53+/+. Durante questo esperimento, hanno anche osservato che l’età mediana di p53+/m era di 96 settimane mentre l’età mediana di p53+/m era di 116-118 settimane., Tyner e colleghi hanno anche esaminato la possibilità che la vita più breve di p53+/m fosse associata all’invecchiamento. Hanno scoperto che dopo 18 mesi, i topi p53+/m hanno iniziato a perdere peso e vigore, come per i topi p53+/m, sono stati osservati pesi ridotti all’età di 30-36 mesi. i topi p53+/m presentano anche lordokyphosis. A seconda dell’analisi a raggi X, i topi p53+ / m hanno mostrato una densità ossea ridotta all’età di 12 mesi e diventeranno gravi all’età di 18 mesi. Questo è un simbolo dell’osteoporosi e l’osteoporosi è un marker dell’invecchiamento negli esseri umani e nei topi . Tyner et al., testato anche la tolleranza allo stress, in quanto questa capacità è anche un marker di invecchiamento . Hanno eseguito biopsie di punzonatura di 3 mm nella pelle posteriore di topi p53+/m e p53+/+ anestetizzati vecchi e giovani. I loro risultati hanno mostrato che molti vecchi topi p53+ / m sono morti dopo aver iniettato la dose standard di Avertin, indicando che i vecchi topi p53+/m erano meno tolleranti allo stress .
Nel 1991, è stato scoperto che p53 ha la capacità di indurre apoptosi, mentre nel 2003; Mihara e altri scienziati hanno scoperto che p53 ha anche un ruolo di apoptosi nei mitocondri ., Poiché alcune proteine mitocondriali hanno la capacità di attivare l’apoptosi cellulare mediante caspasi attive o neutralizzando gli inibitori citosolici. Nell’esempio della caspasi indotta dal citocromo c, dopo aver ricevuto il segnale di apoptosi, il citocromo c viene rilasciato dallo spazio intermembrana dei mitocondri e quindi a sua volta si lega ad Apf-1 che esiste come monomero inattivo, induce il suo cambiamento conformazionale e aumenta la sua affinità di legame per dATP/ATP di 10 volte rispetto a Apaf-1 lega dATP/ATP da solo. Quindi il complesso Apaf-1-citocromo c si lega a dATP / ATP, forma l’apoptosoma., Dopo di che il dominio di reclutamento caspasi (CARTA) di Apaf-1 esposto nell’apoptosoma, reclutare procaspase-9, e quindi autoattivarsi. Il complesso finale quindi fende e attiva altre caspasi come caspasi-3 che a sua volta successivamente fendono importanti molecole nella cellula, causando la condensazione della cromatina, la frammentazione del DNA e infine portando all’apoptosi . La figura 2 mostra la via di attivazione della caspasi indotta dal citocromo C.
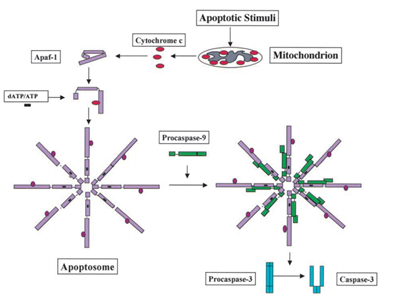
Mihara M et al. gli scienziati hanno scoperto che il gene p53 wild-type può essere traslocato rapidamente sulla superficie mitocondriale delle cellule tumorali. Nell’esperimento, hanno scoperto che alcune proteine p53 wild-type indotte dallo stress hanno la capacità di traslocare nei mitocondri dei timociti nelle cellule umane o di topo dopo l’apoptosi a causa del danno al DNA e dell’ipossia. Quindi queste proteine p53 wild-type inducono la permeabilizzazione dei mitocondri e causano una serie di cambiamenti che si verificano nei mitocondri come il rilascio del citocromo c formando un complesso con Bcl2 e BclXL .,
Come un buon risultato clinico con poco effetto collaterale, la terapia genica è popolare. Entro la fine del 2005, ci sono stati 1020 studi di terapia genica nel database del Journal of Gene Medicine. Tra questi studi, il 66% delle terapie geniche è stato condotto su pazienti oncologici e 58 studi di questo hanno utilizzato rAd-p53, un adenovirus ricombinante che codifica per il gene umano p53. Nell’aprile 2004, un’iniezione umana ricombinante di adenovirus-p53 (Gendicina) è stata lanciata formalmente. Gendicina è usato per trattare il carcinoma a cellule squamose della testa e del collo ed è stato approvato dalla Food and Drug Administration stato della Cina il Ott. 16, 2003., È diventato il primo prodotto di terapia genica nel mondo ad essere approvato dal governo cinese .
Il gene p53 è stato scoperto per regolare il metabolismo nel 2005. Al fine di trasferire dalla fase G1 a S, le cellule devono avere un supporto di materie prime sufficiente per il DNA, gli organelli e la sintesi proteica. Per regolare questo processo, sono necessari alcuni punti di controllo. Uno di questi è il checkpoint glucosio-dipendente a G1/S. È regolato dalla chinasi proteica attivata da AMP (AMPK). Quando il glucosio è esaurito, AMPK può fosforilare la proteina p53, che a sua volta induce l’arresto cellulare ed evita la morte cellulare., Le cellule che incontrano l’arresto p53-dipendente rientreranno nel ciclo cellulare quando il glucosio è sufficiente .
È noto che l’inattivazione di p53 è necessaria per la formazione di tumori. Bykov et al. VJ e Snydel EL et al. sottolinea che il funzionamento improprio di p53 può portare alla proliferazione di un tumore esistente . Ventura e i suoi collaboratori hanno fatto alcuni esperimenti per testare questa ipotesi. Hanno ripristinato la funzione di p53 endogeno nei tumori autoctoni primari per esaminare le conseguenze della riattivazione di p53., Il risultato ha mostrato che la riattivazione di p53 era responsabile della regressione dei tumori autoctoni. Ciò significa che la proteina p53 inattivata può portare allo sviluppo del tumore . Xue e altri scienziati hanno anche fatto un esperimento per testare la conseguenza della riattivazione di p53 sui tumori. Hanno usato reversible RNA interference (RNAi) per regolare l’espressione di p53 endogeno nei topi con cancro al fegato. Nell’esperimento, la doxiciclina (Dox) viene utilizzata per riattivare p53, poiché l’espressione di p53 viene totalmente soppressa quando manca Dox e rapidamente ripristinata quando viene aggiunto Dox., Quando trattato con Dox, p53 miRNA è stato spento, il che a sua volta causa un aumento dell’espressione di p53. Il risultato ha mostrato che i tumori nei topi trattati con Dox diventano non rilevabili dopo 12 giorni, mentre i tumori nei topi non trattati sono cresciuti rapidamente. Per testare la conseguenza della riattivazione transitoria di p53, hanno trattato i topi con Dox per 4 giorni e poi si sono fermati. Il risultato ha mostrato che anche un trattamento di due giorni può causare la regressione dei tumori e 4 giorni di trattamento possono far regredire completamente i tumori., Hanno anche sottolineato che durante la regressione tumorale, la p53 riattivata transitoriamente può innescare la senescenza cellulare, non l’apoptosi. Nello stesso anno, Hu ha scoperto che l’impianto embrionale in topi p53-/- femmina è regolato dal fattore inibitorio della leucemia (LIF). La LIF è una citochina secreta ed è importante per l’impianto di blastocisti. Il gene che codifica LIF è identificato come gene bersaglio p53 e il sito di legame p53 si trova nell’introne 1 sia negli esseri umani che nei topi .