遺伝子p53は1979年に最初に発見されました。 タンパク質は、抗T血清と免疫沈降によってsimianウイルス40形質転換マウス細胞(SV40)で同定された;このタンパク質は、タンパク質p53と呼ばれていた。 同じ年に、Kressと他の科学者は、分子量が50-60kDaの新しいクラスのタンパク質を発見しました。 この種類の蛋白質はp53としてそれから識別されました。 タンパク質p53はまた、免疫沈降によって様々な形質転換細胞株から同定することができる。, レーンとリンツァーも1979年に同様の結果を得た。 P53を同定するための他の証拠は、p53が試験されたすべての形質転換マウス細胞において発現されたことであり、これらの試験には化学誘発肉腫、形質転換線維芽細胞、および白血病が含まれるが、正常細胞ではp53は発現されなかった。 さらに、p53の高レベルは、細胞が自発的または非自発的に、どのように変換されたかに関係なく、ほとんどの形質転換細胞で検出されませんでした。, それは主にp53の安定性の増加によるものであったが、F9胚性カルチノア細胞において、高レベルのp53を発現し、これは翻訳されたp53mRNAの量に
タンパク質p53が1979年に発見された後、それを分析することが一般的になりました。 しかし、当時、それは新たに発見されたタンパク質であり、それには以前の名前がなかったため、異なる機関が異なる名前を使用し、異なる名前の論文を, この問題を解決するために、1983年に、英国Oxtedで開催された第1回国際p53ワークショップでは、異なる国の異なる研究グループの科学者が集まり、この新しく発見されたタンパク質の共通の命名法について議論しました。 この会議では、”p53″がその名前となり、それ以来使用されています。 科学者がタンパク質p53と呼んだ理由は、このタンパク質の分子量がSDSゲル中のその移動に基づいている53kDaであるためであると考えられていた。 その後、分子量は間違っていることが証明され、正しい分子量は43でなければならない。,7kDa p53にはプロリンに富む領域が含まれており、この領域はSDSゲル中のp53の移動を減らすことができるためです。 しかし、”p53″という名前は残っていました。
1980年代、タンパク質p53は細胞周期に関与し、DNA複製に役割を果たしていると考えられていた。 その後、1982年から1994年にかけて、人々はいくつかのウイルス性腫瘍蛋白質がp53に結合して複合体を形成することができることを発見した。 1982年、Sarnow et al. アデノウイルスE1b(58kDa)は、上記の54KDAタンパク質SV40形質転換マウス細胞に存在すると相互作用することができることがわかった。, T抗体の免疫学的特異性および54kDaタンパク質のペプチドマップの結果によれば、この54kDaタンパク質はp53と同定される。 同じ年に、科学者たちは、彼らがスイスの3T3マウス細胞にp53抗体を注入した場合、それは細胞周期のS期に入る細胞を阻害することがわかったが、同じ状況下で、p53抗体はSV40またはアデノウイルス誘導DNA合成に影響を与えなかった。,
1984年の後半に、科学者たちは、非変換された3t3線維芽細胞に対するp53の効果を調べ、異なる時点でのタンパク質p53の合成速度を分析し、G1期 この結果は、タンパク質p53が間期から分裂期に入る細胞を阻害することを示唆している。 Maltzman W et al. 同じ年に別の実験を行いました。 彼らは、uv光とUV擬態化学発癌物質4NQOで非変換マウス細胞を処理し、彼らはp53の高レベルを検出しました。, その結果、p53の発現の上昇は、細胞周期を示すシンボルであるだけでなく、DNA合成および細胞増殖に関与する成分でもあることが示された。 1987年、シミアンウイルス40とDNAポリメラーゼαのT抗原の複合体を研究したとき、Gannonなどの科学者は、p53とポリメラーゼαに結合したときに抗原に同様の変化を見出した。 彼らはまた、三つの成分の特定の濃度で、T抗原、p53およびDNAポリメラーゼαを含む特別な三量体複合体を形成することができることを見出した。, T抗原はウイルスDNA複製および細胞形質転換に関与しているので、この結果は、p53が細胞周期およびDNA複製の制御において役割を果たすことを示
実験が上で示したように、p53は細胞を不死化する能力を有する。 1984年、Eliyahu D et al. p53および癌遺伝子mycの生成物は、いくつかの同様の特性を共有することがわかった。 それらの両方は、他のタンパク質に結合する能力を有し、細胞周期に関与し、そしてそれらは両方とも形質転換細胞の核に蓄積する。, Bienz、Pennica、Orenは、タンパク質p53とmycの生成物のアミノ酸配列を分析し、二つのタンパク質が分子構造と特別な荷電残基の位置に類似点を示すことを見出した。 その後、科学者たちは、p53が癌遺伝子として作用する可能性があることを提案した。 この仮説に基づいて、Eliyahu D et al. いくつかの実験をした。 一次率胚線維芽細胞は、myc産物とHa-rasの両方の関与によって形質転換することができるので、一次赤ちゃんラット腎臓細胞は、Ha-rasとアデノウイルス初期領域1Aの協力によって形質転換することもできる、Eliyahu D et al., この種の生物学的試験システムを使用して、p53の発癌機能を同定することに決めた。 彼らはp53と正常な胚細胞を処理し、Ha-rasを活性化した。 その結果、標的細胞は形態変化に遭遇し、高レベルのp53を産生することが示された、Eliyahu D et al. p53およびHa-rasによる胚性線維芽細胞の形質転換は、遺伝子p53が癌遺伝子であることを説明したと考えられている。, 1985年、ジェンキンスは、p53遺伝子が細胞の寿命を延ばし、安定したタンパク質の産生を引き起こす可能性のあるコード配列を再配置することによって形質転換の感受性を高めることができることを提案した。
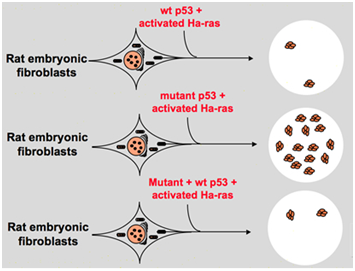
しかし、1980年代後半に、科学者たちは、p53が癌遺伝子の代わりに腫瘍抑制遺伝子であることを認識し始めました。 彼らは、正常な機能を持つp53が腫瘍の多くで検出できないことを観察し、野生型p53遺伝子の発現および機能を失うことが細胞形質転換の間に必, これらは、野生型p53遺伝子が新生物の進行を阻害する可能性を高める。 その後、彼らは別の仮説を定式化した:以前の実験で使用されたクローン遺伝子p53は、時折非常に保存されたドメイン内の支配的な負の変異を含み、反対の実験結果につながる。 1988年に、ベンおよび他の科学者はマウスの赤白血病細胞ラインの再配列されたp53を検出しました–友人の白血病のウイルスに感染するマウスの脾臓から得られるDP20-1およびCB3。, 1989年に、P53が癌遺伝子であることを指摘したEliyahuは彼の心を変え、野生型p53遺伝子が細胞形質転換を阻害する可能性があると考えました。 Eliyahuと他の科学者は、in vitroで様々な癌遺伝子の組み合わせによって一次率胚線維芽細胞変換を誘発する能力にプラスミドおよび変異体p53によってコードされた野生型p53タンパク質の効果を研究しました。 例えば、突然変異体p53plus ras、およびmyc plus ras。, その結果、野生型p53は変異p53プラスrasによって引き起こされる変換された病巣の巨大な減少につながることを示した;変異p53はmycプラスras媒介変換は、野生型p53の発現に非常に敏感であるが、mycプラスrasによって引き起こされる変換された病巣に阻害を示さなかった。 図1は、この実験を簡潔に示しています。 変異体p53と比較して、野生型p53は細胞形質転換に対して明らかに阻害効果を示すことを示した。 この効果は、野生型p53の発現レベルに正の関連があり、突然変異体p53の発現レベルに負の関連がある。, この実験は、野生型p53が実際に変異p53と比較して反対の機能を有し、腫瘍形成を阻害する可能性があることを示唆した。 現在、p53は腫瘍抑制遺伝子として認識されている。 腫瘍の約半分がp53によって引き起こされると推定されている。 これは、ヒトで最も頻繁に変異した遺伝子の一つであり、世界中で最も頻繁に分析された遺伝子です。
1980年代の最初の数年間、p53の生化学的経路およびp53変異の影響は明らかではなかった。 1991年、Kernらの科学者たちは、33塩基対DNA配列がin vitroで野生型p53に特異的に結合することを発見しました。 彼らはまた、p53タンパク質には、この特定のDNA領域に結合できないヒト腫瘍に通常見られる二つの突然変異が含まれていることを発見した。 そこで彼らは、p53の機能は特定のDNA配列に結合する能力に依存し、この能力はヒト腫瘍に見られる突然変異によって変化すると考えました。, 彼らはまた、この33塩基対DNA配列が、ヒトのp53に特異的に結合する能力を有する唯一の配列ではないかもしれないと仮定しているが、p53の機能をよりよく理解するのに役立つ可能性がある。 後で、p53は細胞周期、DNA修理、apoptosisおよびangiogenesisを始める微分の間に役割を担うために見つけられました。 ロッター Vら。 このp53上が制御する分化細胞である。 例えば、高レベルのp53タンパク質は、B細胞分化中にいくつかの重要なステップで検出された。 上昇したp53はまた、精子形成中に検出することができる。, 一方、成体マウスのいくつかの器官では、非常に低レベルのp53タンパク質のみが検出され得る。
1990年には、有用なツールが時折発見されました。 これは、p53val135と呼ばれるp53の温度感受性変異体である。 それは32.5ocの温度で実質の野生タイプp53として機能できま、変形を抑制し、また他の変異させたp53として37.5ocの温度または48ocの上で機能でき、変形を引き出します。 さらに、p53val135を発現する形質転換細胞については、その増殖は許容温度で制御され、この種の制御は可逆的である。, このp53val135変異体を用いることにより、野生型p53はG1またはG2/Mのいずれかで細胞周期停止を誘導することが発見された。 1991年、Elisheva et al. 温度感受性p53val135は、マウス骨髄性白血病細胞株において異なる機能を果たすことがわかった。 数日間p53val135の再活性化後、すべての細胞が死亡し、この死はアポトーシスのいくつかの特性を示す。 一年後、同様の結果がShawによって得られました。 野生型p53は、ヒト結腸腫瘍由来細胞株EBにトランスフェクトされました。, 細胞を光顕および電子顕微鏡下で調べ,アポトーシスのいくつかの特性を示すことが分かった。 1990年、Scheffner et al. そして、他の科学者は、宿主細胞調節タンパク質の破壊を刺激するE6は、発癌性ヒトパピローマウイルス16型および18型によってコードされ、それが順番にタンパク質p53の分解を引き起こすin vitroで野生型p53との複合体を形成することができることを発見しました。
1992年に、重要なタンパク質MDM2がp53と緊密に結合し、p53によって媒介されるトランス活性化を阻害することから発見された。, MDM2の分子量は90kDaであり、突然変異および野生型p53の両方と複合体を形成する。 同じ年に、Livingstone RL et al. 細胞が野生型p53対立遺伝子の一方または両方のコピーを失ったかどうか、およびそれが遺伝子増幅を引き起こすのに十分であるかどうかを調べた。 遺伝子増幅は形質転換細胞ではほとんど検出されたが,正常線維芽細胞では検出されなかった。 その結果、p53対立遺伝子の一方のコピーを失った細胞は野生型p53として作用し、野生型p53対立遺伝子の両方のコピーを失った細胞はより高い増 Yin Yらによって行われた別の実験。, 同様の結果を示した。
1993年に、CDKN1Aと呼ばれるp53標的遺伝子が同定された。 これは、サイクリン依存性キナーゼ阻害剤であるタンパク質p21をコードし、サイクリン-CDK2およびCDK1をそれらに結合することによって阻害する。 1993年、SzekelyはEpstein-Barrウイルス核抗原5(EBNA-5)がEpstein-Barrウイルスによってコードされており、ヒトBリンパ芽球様細胞に感染することができることを発見した。 66アミノ酸長いペプチドは複雑なEBNA-5-p53の形成に責任があり、p53の点突然変異はEBNA-5への結合能力に影響を与えなかった。, しかしながら、それは他の分子との複合体の形成を阻害する。 1994年、Choと彼の同僚は、複雑なp53-DNAの結晶構造を最初に記述しました。 このDNA結合ドメインはコアドメインとも呼ばれた。 それは残基102-292を含み、ベータサンドイッチからなる。 また、コアドメインの詳細な構造を実証しました。 また、1994年には、Wang XW et al. ヒトにおけるb型肝炎ウイルスXタンパク質(HBX)と野生型p53タンパク質との相互作用。, 彼らは、HBXがp53に結合した後、P53が他の配列特the DNAに結合する能力を阻害し、p53と転写因子との会合を阻害することもできることを見出した。
1997年、Honda R et al. まずMDM2p53ユビキチン化をトリガーし、ユビキチンプロテアソームシステムによってp53の分解につながることができると仮定しました。 彼らは、MDM2がp53のN末端ドメイン(NTD)に結合し、ユビキチンリガーゼE3として作用することを指摘した。 また、1997年には、p63とp73の二つの新しいタンパク質ファミリーが発見され、p53と実質的な相同性を共有している。, 腫瘍タンパク質73とも呼ばれるp73は、1p36に位置する遺伝子によってコードされる。 その場所は神経芽細胞腫および他の腫瘍で頻繁に削除される。 p73は、p53標的遺伝子を活性化し、p53と相互作用することができる。 Yang et al. 遺伝子p63が3q27-29に位置し、様々なマウスおよびヒト細胞で検出することができることがわかった。 P73のように、p63はまたapoptosisを引き起こすことができますp53ターゲット遺伝子をかなりtransactivateできます P63の特徴の一つは、p63の大部分がN末端を欠いていることである。, 同じ年に、Serranoおよび共同研究者は、p53またはp16の非存在下でrasによって一次マウス線維芽細胞を形質転換することができ、不活性なp53またはp16がヒト細胞の不死化プロセスを促進することができることを見出した。 これらの知見は、p53が細胞老化に役割を果たしていることを示唆している。 それから、1997年に、p53はapoptosisの開始の役割を担うために見つけられました。 細胞が増殖期に入ると、各染色体の終わりにあるテロメアは、DNAスタンドの終わりにある一本鎖DNAの不完全な複製のために、DNA複製の各ラウンドの後, 活性化された腫瘍抑制遺伝子p53は、細胞分裂が起こり得る回数を制限する。 ウィンフォードTDは、野生型p53の機能の損失と、すべての線維芽細胞は、アポトーシスから脱出することがわかりました。 また、p53のトランス活性化機能はアポトーシスによってオンにすることができる。 Wynford TDは、p53がどのように活性化されるかについて三つの可能性があると提案した。 最初のものはリン酸化による翻訳後修飾であり、第二のものはp33ING1のような転写補因子をアップレギュレーションし、最後のものはMDM2のようなp53阻害剤をダウンレギュレーションしている。
2000年、Brodsky MH et al., ショウジョウバエにおけるp53の転写標的を研究した。 ショウジョウバエの目がヒトp53の発現の下で重度の粗い目の表現型を示すことを示す証拠があり、眼の成虫円板細胞のアポトーシスを誘導し、色素細胞の喪失を引き起こし、最終的にショウジョウバエの眼の発達を阻害するので、ショウジョウバエはp53の機能を研究するためのモデル動物である可能性がある。 ブロツキーは、遺伝子rprがrprのcis調節領域に位置するコンセンサスp53結合部位を含み、それもアポトーシスの活性化因子であることを見出した。, 他の証拠とともに、ブロツキーはrprがp53の転写標的の一つであると主張した。 2001年、Derryらは、c.elegansにはp53遺伝子はないが、実際にはタンパク質p53と同様の配列を持つタンパク質をコードする遺伝子cep-1が含まれていることを発見した。 このc.elegans遺伝子は遺伝毒性ストレスによってアポトーシスを誘導する能力を有し、減数分裂の間に必要なコンポーネントであるタンパク質CEP-1をコード
2002年、Tynerと共同研究者は、p53が生物の老化を調節する役割を果たすことを提案した。, P53の機能を研究するために、彼らはエクソン1-6とp53+/mと呼ばれる野生型p53遺伝子(p53+/+)の上流領域を削除することにより、変異p53を有する遺伝子工学的マウスを作成し、野生型p53として作用し、野生型p53よりも自然発生腫瘍に対する耐性を高めた。 実験では、p53+/m、p53+/+およびp53+/-を含むマウスを監視した。 p53+/-は、野生型p53遺伝子の一つのコピーを失うマウスを意味する。, 結果は、p53+/mを有するマウスのいずれも生命を脅かす腫瘍を開発しなかったことを示したが、p53+/-を有するマウスの80%以上およびp53+/+を有するマウスの45%以上がこれらの種類の腫瘍を開発した。 腫瘍の内部を見ると、ローカライズされた腫瘍病変は、2 35p53+/mマウスのうち、対照的に、リンパ腫のような様々な腫瘍で観察され、骨肉腫はp53+/-およびp53+/+ この実験の間、彼らはまた、p53+/mの年齢中央値が96週間であったのに対し、p53+/mの年齢中央値が116-118週間であることを観察した。, タイナーと同僚はまた、p53+/mの短い寿命が老化に関連していた可能性を検討した。 彼らは、18ヶ月後、p53+/mマウスは体重と活力を失い始め、p53+/mマウスは30-36ヶ月の年齢で体重の減少が観察されたことを発見した。 p53+/mマウスはまた、前弯後弯症を示す。 X線分析によっては、p53+/mマウスは12ヶ月の年齢で骨密度の低下を示し、18ヶ月の年齢で重度になるでしょう。 これはosteoporosisしょう症の象徴であり、osteoporosisしょう症はヒトおよびマウスの老化のマーカーである。 Tyner et al., また、この能力も老化のマーカーであるため、ストレスの耐性をテストしました。 彼らは、老いも若い麻酔p53+/mおよびp53+/+マウスの後部皮膚に3mmパンチ生検を行った。 彼らの結果は、古いp53+/mマウスの多くは、古いp53+/mマウスがストレスにあまり耐性であったことを示す、Avertinの注入標準用量後に死亡したことを示した。
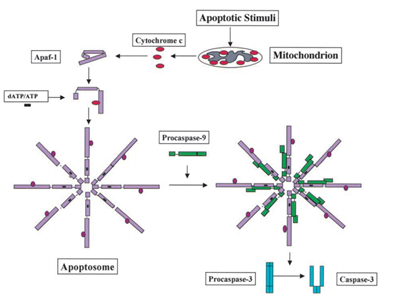
1991年には、p53はアポトーシスを誘導する能力を有することが判明し、2003年には、三原らの科学者は、p53はまた、ミトコンドリアにおけるアポトーシスの役割を持っていることが判明しました。, いくつかのミトコンドリアタンパク質は、活性カスパーゼまたは細胞質ゾル阻害剤を中和することによって細胞アポトーシスを活性化する能力を シトクロムc誘導カスパーゼの例では、アポトーシスシグナルを受け取った後、ミトコンドリアの膜間空間からシトクロムcが放出され、不活性モノマーとして存在するApf-1に結合し、その立体配座変化を誘導し、Apaf-1がdATP/ATP単独に結合するよりもdATP/ATPに対する結合親和性を10倍に増加させる。 その後、複合体Apaf-1-シトクロムcはdATP/ATPに結合し、アポトソームを形成する。, その後、Apaf-1のカスパーゼ募集ドメイン(CARD)は、アポトソームに露出し、procaspase-9を募集し、その後、自分自身を自動活性化します。 最終的な複合体はそれからそれから続いてクロマチンの凝縮、DNAの分裂を引き起こし、最終的にapoptosisの原因となるセルの重要な分子を切断するcaspase-3のような他のカスパーゼを切断し、活動化させます。 図2は、シトクロムcによるカスパーゼ活性化経路を示しています。
Mihara M et al. 科学者たちは、野生型p53遺伝子が急速に腫瘍細胞のミトコンドリア表面に移動することができることを発見しました。 実験では、彼らは、いくつかのストレス誘発性野生型p53タンパク質は、DNA損傷および低酸素症によるアポトーシス後にヒトまたはマウス細胞の胸腺細胞のミトコンドリアに移動する能力を有することを見出した。 その後、これらの野生型p53タンパク質は、ミトコンドリアの透過を誘導し、Bcl2およびBclXLとの複合体を形成することによってシトクロムcを放出するようなミトコンドリアで起こる一連の変化を引き起こす。,副作用の少ない良好な臨床成績として,遺伝子治療が普及している。 2005年末までに、Journal of Gene Medicineのデータベースには1020件の遺伝子治療の試験がありました。 これらの試験のうち、遺伝子治療の66%が癌患者に対して実施され、58の試験では、ヒトp53遺伝子をコードする組換えアデノウイルスであるrAd-p53が使用された。 2004年、組換えヒトアデノウイルス-p53注射(ジェンディシン)が正式に開始された。 Gendicineがヘッドを扱うのに使用され、首の扁平上皮癌およびOctの中国の州の食品医薬品局によって承認されました。 16, 2003., それは中国の政府によって承認されるべき世界の最初の遺伝子療法プロダクトになりました。
遺伝子p53は2005年に代謝を調節することが発見された。 G1からS期に移行するためには、細胞はDNA、細胞小器官およびタンパク質合成のための十分な原料サポートを有していなければならない。 を規制するこのプロセスは、検問所が必要となります。 そのうちの一つはG1/Sのグルコース依存性チェックポイントであり、AMP活性化プロテインキナーゼ(AMPK)によって調節される。 グルコースが排出されると、AMPKはタンパク質p53をリン酸化することができ、これは細胞停止を誘導し、細胞死を回避する。, P53依存性停止に遭遇する細胞は、グルコースが十分であるときに細胞周期を再入力する。
腫瘍の形成にはp53の不活性化が必要であることが知られている。 Bykov et al. VjおよびSnydel EL et al. p53の不適切な機能が既存の腫瘍の増殖につながる可能性があることを指摘する。 Venturaと彼の同僚は、この仮説をテストするためにいくつかの実験を行った。 彼らは、p53再活性化の結果を調べるために、原発性自在性腫瘍における内因性p53の機能を回復した。, その結果、p53再活性化が自生腫瘍の退行の原因であることが示された。 その不活性化はp53タンパク質で腫瘍を開発。 Xueと他の科学者はまた、腫瘍に対するp53の再活性化の結果をテストするための実験を行った。 彼らは、肝臓がんを有するマウスにおける内因性p53の発現を調節するために可逆RNA干渉(RNAi)を使用した。 実験では、doxが欠落しているときにp53の発現が完全に抑制され、Doxが添加されると急速に回復するので、p53を再活性化するためにドキシサイクリン(Dox)を使用している。, Doxで処理すると、p53miRNAが遮断され、p53の発現増加が引き起こされた。 その結果、dox処理マウスの腫瘍は12日後に検出できなくなり、未処理マウスの腫瘍は急速に成長したことが示された。 P53の一時的な再活性化の結果をテストするために、彼らは4日間Doxでマウスを処理し、その後停止しました。 その結果、二日間の治療でさえ腫瘍の退行を引き起こす可能性があり、4日間の治療で腫瘍が完全に退行する可能性があることが示された。, 彼らはまた、腫瘍退縮中に、一時的に再活性化されたp53がアポトーシスではなく細胞老化を引き起こす可能性があることを指摘した。 同じ年に、Huは、p53-/-雌マウスにおける胚移植が白血病抑制因子(LIF)によって調節されることを見出した。 LIFは分泌されたサイトカインであり,はい盤胞移植に重要である。 LIFをコードする遺伝子はp53ターゲット遺伝子として識別され、p53結合部位は人間およびマウス両方のイントロン1にあります。