zautomatyzowana procedura napisana w Microsoft Visual Basic (VB) 6.,0 aktualizuje Davida co tydzień za pomocą następujących procedur: wywołanie serii aplikacji Perl i Java, które pobierają dane publiczne za pośrednictwem protokołów anonimowego transferu plików (FTP) (Tabela 1); Rozpakowanie i przetworzenie żądanych danych adnotacji; tworzenie rozdzielanych tabulatorami plików danych gotowych do importu bazy danych; i import danych do systemu zarządzania relacyjnymi bazami danych Oracle 8i (RDBMS) przy użyciu aplikacji Oracle SQL*Loader. Microsoft IIE web server i technologia Active Server Page są używane do uzyskania dostępu do bazy danych za pomocą JavaBeans i structured Query language (SQL)., Numery LocusLink dla zestawów sond Affymetrix pochodzą od stowarzyszeń University of Michigan lub NetAffx . Adnotacje funkcjonalne i odsyłacze baz danych pochodzą z LocusLink, który zapewnia stabilne, reprezentowane przez człowieka reprezentacje genów. Więcej szczegółowych informacji na temat źródeł danych używanych przez Davida można znaleźć w sekcji FAQ na stronie .,
Moduły analizy
DAVID składa się z czterech głównych modułów: narzędzia do adnotacji, GoCharts, KeggCharts i DomainCharts. Narzędzie do adnotacji jest zautomatyzowaną metodą funkcjonalnej adnotacji list genów. Dowolną kombinację danych adnotacji można wybrać spośród 10 opcji, zaznaczając odpowiednie pola wyboru (Tabela 2)., Adnotacje są dodawane do listy przesłanych genów poprzez wybranie przycisku upload, który zwraca tabelę HTML zawierającą oryginalną listę identyfikatorów użytkownika dołączoną do wybranych adnotacji funkcjonalnych. Nieoznakowane geny są włączane do wyjścia bez dołączonych danych do celów śledzenia.,
moduł GoCharts graficznie wyświetla rozkład genów wyrażonych różnie wśród kategorii funkcjonalnych przy użyciu kontrolowanego słownictwa Konsorcjum ontologii genów (GO), które zapewnia strukturalny język, który może być stosowany do Funkcje genów i białek we wszystkich organizmach, nawet gdy wiedza nadal gromadzi się i zmienia ., Język jest skonstruowany w ukierunkowanym grafie acyklicznym (DAG), w którym specyficzność terminowa wzrasta, a pokrycie genomu maleje wraz z przesuwaniem się w dół hierarchii. W przeciwieństwie do prawdziwej hierarchii, terminy dziecięce w DAG mogą mieć więcej niż jeden termin rodzica i mogą mieć inną klasę relacji z różnymi rodzicami. Struktura GO zaczyna się od trzech głównych kategorii, procesu biologicznego, funkcji Molekularnej i składnika komórkowego., Proces biologiczny obejmuje szerokie cele biologiczne, takie jak mitoza lub metabolizm puryn, które są realizowane przez uporządkowane zespoły funkcji molekularnych. Funkcja molekularna opisuje zadania wykonywane przez poszczególne produkty genów; przykładami są czynnik transkrypcyjny i helikaza DNA. Typ klasyfikacji komponentów komórkowych obejmuje struktury subkomórkowe, lokalizacje i kompleksy makrocząsteczkowe; przykłady obejmują jądro, telomer i Kompleks rozpoznawania pochodzenia., Po wybraniu typu klasyfikacji poziomy określające zasięg i specyfikę listy są wybierane przez wybranie odpowiedniego przycisku radiowego. Poziom 1 zapewnia najwyższy zasięg listy z najmniejszą ilością specyficzności terminowej. Z każdym rosnącym poziomem pokrycia maleje, podczas gdy specyficzność wzrasta tak, że poziom 5 zapewnia najmniejszą ilość pokrycia o najwyższej specyficzności term.
dane klasyfikacyjne są wyświetlane jako wykres słupkowy, gdzie długość słupka reprezentuje liczbę identyfikatorów genów w każdej kategorii., Użytkownik może ustawić parametry wizualizacji do sortowania danych wyjściowych i wyświetlania kategorii, które zawierają co najmniej minimalną liczbę genów. Wybranie pojedynczego paska otwiera nową tabelę HTML wyświetlającą identyfikator genu, numer LocusLink, nazwę genu, aktualną klasyfikację i inne klasyfikacje dla każdego genu w tej kategorii. Przycisk” Pokaż wszystkie „otwiera nową tabelę HTML wyświetlającą wszystkie dane klasyfikacji, a przycisk” Pokaż Dane wykresu ” otwiera tabelę HTML zawierającą podstawowe dane wykresu, umożliwiając użytkownikom odtworzenie niestandardowej Grafiki wykresu w programie arkusza kalkulacyjnego., Nowy wykres może być wyświetlany dla dowolnej podgrupy genów, wybierając typ klasyfikacji i poziom za pomocą pól wyboru i przycisków opcji dostępnych na bieżącej stronie Użytkownika, które pozwalają na drill-down możliwości. Liczba genów opatrzonych adnotacją jest uwzględniana w wyjściu, a geny nie opatrzone adnotacją są przypisywane do kategorii „niesklasyfikowane”, co zapewnia użytkownikom automatyczny system śledzenia genów nie opatrzonych adnotacją.
KeggCharts graficznie przedstawia rozkład genów wyrażonych różnie wśród szlaków biochemicznych KEGG., Każda ścieżka jest powiązana z mapą szlaku KEGG, na której geny o różnej ekspresji z oryginalnej listy są zaznaczone na Czerwono. W tym ujęciu geny są dodatkowo powiązane z dodatkowymi adnotacjami dostępnymi za pośrednictwem systemu pobierania DBGET KEGG . Podobnie jak w przypadku GoCharts, użytkownik może ustawić parametry wizualizacji dla sortowania danych wyjściowych i wyświetlania kategorii, które zawierają co najmniej minimalną liczbę genów, a wizualizacja KeggCharts dziedziczy wszystkie dynamiczne cechy GoCharts.
DomainCharts wyświetlają rozkład genów wyrażonych różnie wśród domen białka PFAM ., Każda nazwa domeny jest powiązana z zachowaną bazą domen (CDD) Narodowego Centrum Informacji biotechnologicznej (NCBI), gdzie szczegóły dotyczące funkcji domeny, struktury i sekwencji są łatwo dostępne. Podobnie jak w przypadku GoCharts i KeggCharts, użytkownik może ustawić parametry wizualizacji dla sortowania danych wyjściowych i wyświetlania kategorii, które zawierają co najmniej minimalną liczbę genów, a Wizualizacja DomainCharts dziedziczy wszystkie dynamiczne cechy GoCharts i KeggCharts. Aby uzyskać więcej informacji na temat funkcjonalności DAVID odwiedź sekcję FAQ na stronie .,
Korzystanie DAVID do kopalni funkcjonalne adnotacji
aby zademonstrować funkcjonalność DAVID przeanalizowaliśmy listę genów differentially expressed in human peripheral blood mononuclear cells (PBMC) po inkubacji z HIV-1 otoczki białek. Szczegóły eksperymentu, przygotowanie RNA, i GeneChip hybrydyzacji procedur, wraz ze szczegółami chip-to-chip normalizacji i statystycznej analizy różnicowej ekspresji genów są dostarczane w Cicala et al. ., Krótko, pierwotne ludzkie PBMC i makrofagi pochodzące z monocytów były inkubowane przez 16 godzin z białkiem otoczki HIV-1 (gp120). Mikrocząsteczki oligonukleotydów o wysokiej gęstości (Affymetrix HU-95A GeneChip) były używane do monitorowania zdarzeń transkrypcyjnych indukowanych przez gp120. W wyniku tej analizy zidentyfikowano 402 geny o różnej ekspresji.
podczas gdy 16 genów modulowanych przez HIV-1 gp120 było wcześniej związanych z replikacją HIV i/lub sygnalizacją otoczki, Pozostałe geny mają nieznaną funkcję lub nigdy nie były związane z HIV-1 lub gp120., Przekształcenie tej listy genów w znaczenie biologiczne wymaga zebrania istotnych informacji z kilku repozytoriów danych. Dla wielu badaczy proces ten polega na iteracyjnym przeglądaniu kilku baz danych dla każdego genu, ręcznym zbieraniu informacji specyficznych dla genów dotyczących sekwencji, funkcji, ścieżki i Stowarzyszenia Chorób. W przeciwieństwie do tego, systematyczne podejście Dawida jednocześnie dodaje biologicznie bogate informacje pochodzące z kilku publicznych źródeł danych do list genów równolegle., Wybranie narzędzia adnotacji Davida i przesłanie listy 402 różnie wyrażonych genów inicjuje funkcjonalną adnotację i analizę całego zbioru danych. Po przesłaniu lista genów jest przechowywana przez całą sesję analizy, umożliwiając użytkownikom przełączanie się między modułami bez konieczności ponownego przesyłania danych.
narzędzie do adnotacji
narzędzie do adnotacji udostępnia kilka opcji adnotacji i tworzy tabelaryczny widok listy genów użytkowników i dostępnych adnotacji (Tabela 2)., Wybranie pól adnotacji symbol genu, LocusLink, OMIM, Unigene, Sekwencja referencyjna i nazwa genu, a następnie wybranie przycisku „Prześlij”, tworzy tabelę HTML w przeglądarce internetowej zawierającą wszystkie geny i dostępne do nich adnotacje, gdzie identyfikatory genów, dane opisowe i klasyfikacyjne są pobierane z bazy danych i dołączane do listy genów (Rysunek 1). Identyfikatory genów, takie jak symbol genu i LocusLink, są hiperłączami do dodatkowych danych specyficznych dla genów dostępnych w ich oryginalnych źródłach, zapewniając w ten sposób dogłębne szczegóły specyficzne dla genów i rodowody adnotacji., Dane klasyfikacyjne i podsumowania funkcjonalne mogą być używane do szybkiego skanowania w poszukiwaniu informacji istotnych dla eksperymentalnego systemu badacza. Czas serwera wymagany do wykonania tego modułu koreluje liniowo z wielkością listy genów i zajmuje mniej niż 45 sekund dla list do 1000 genów (Rysunek 2, liczby w nawiasach przedstawiają wartości r2). Wyniki te pokazują moc i wydajność zintegrowanego podejścia do funkcjonalnej adnotacji dużych zbiorów danych.,

udostępnienie narzędzia do adnotacji. Pokazane są dołączone adnotacje dla pierwszych kilku zestawów sondy Affymetrix w tabeli HTML zawierającej wszystkie 402 wpisy. Kategoryczne informacje o warunkach eksperymentalnych zostały przekazane wraz z identyfikatorami zestawu sond Affymetrix i włączone do wyjścia w kolumnie Wartość. Identyfikatory takie jak Symbol, locuslink, OMIM, RefSeq i unigene są hiperłączami do źródeł ich pochodzenia w celu uzyskania bardziej szczegółowych informacji., Tekst zawarty w polach sumarycznych pochodzi z opisowych, funkcjonalnych informacji zawartych w raportach LocusLink opracowanych przez NCBI.
analiza czasu Narzędzia do adnotacji. Czas serwera wymagany (Oś y) do jednoczesnego dołączania wszystkich 10 opcji adnotacji do list genów o rozmiarze od 100 do 1000 (oś x)., Średnia z trzech badań dla list genów zawierających identyfikatory Affymetrix, GenBank, LocusLink i UniGene jest pokazana, a liczby w nawiasach reprezentują wartość R2 korelacji między rozmiarem listy genów a czasem serwera wymaganym do adnotacji.
GoCharts
wybór modułu GoCharts otwiera nowe okno z różnymi opcjami., Użytkownicy wybierają między trzema ogólnymi typami klasyfikacji (proces biologiczny, funkcja molekularna i komponent komórkowy) i pięcioma poziomami adnotacji, które reprezentują zakres terminów i specyficzność (patrz sekcja Moduły analizy). Można określić dowolną kombinację klasyfikacji i poziomu pokrycia. Dostępne są również opcje adnotacji list genów ze wszystkimi dostępnymi terminami GO lub tylko najbardziej szczegółowymi terminami, które są określane jako węzły końcowe., Możliwość wyboru różnych poziomów specyficzności terminowej zapewnia potrzebną elastyczność, a tym samym pozwala badaczom dynamicznie określić, który poziom zasięgu i specyficzności najlepiej pasuje do ich danych i etapu analizy. Na przykład, wczesna faza analizy może składać się z adnotacji genów listy z bardzo ogólnymi terminami w celu uzyskania szerokiego zrozumienia danych. W tym przypadku wybór procesu biologicznego i poziomu 1 klasyfikuje geny za pomocą ogólnych terminów, takich jak” śmierć „i”komunikacja komórkowa”., Zastosowanie zwiększonej specyficzności terminowej ułatwia wydobycie bardziej szczegółowych informacji funkcjonalnych. W tym przypadku selekcja procesu biologicznego i poziomu 5 klasyfikuje geny za pomocą terminów takich jak” apoptotyczne zmiany mitochondrialne „i”percepcja chemosensoryczna”.
jednak zwiększona specyficzność terminowa wiąże się z kosztami, ponieważ wraz ze wzrostem zasięgu listy zmniejsza się (rysunek 3). W naszych badaniach stwierdzamy, że poziom 2 zazwyczaj utrzymuje dobry zasięg, a jednocześnie zapewnia znaczącą specyfikę terminów., Rysunek 4a ilustruje, w jaki sposób wizualizacja Gochartsa szybko ujawnia, że 35 różnie wyrażonych genów bierze udział w”reakcjach stresowych”. Każdy termin GO może być wyświetlany w widoku drzewa lub DAG przez hiperłącza do QuickGO .
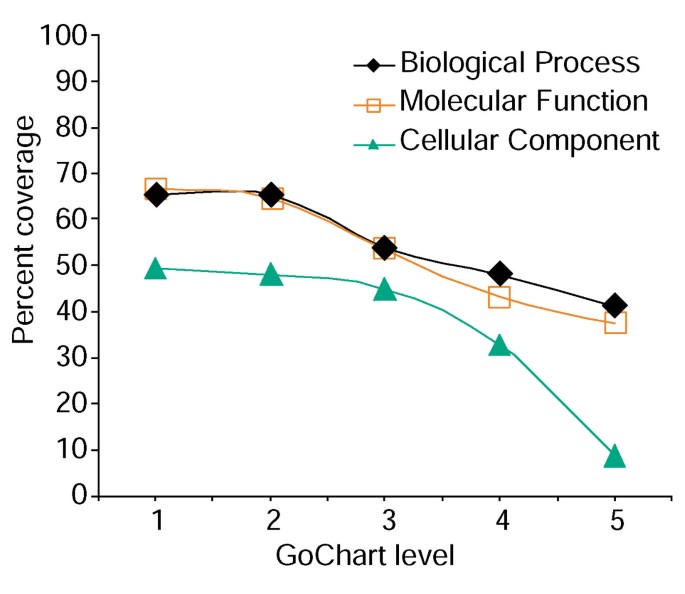
Analiza pokrycia listy genów przy użyciu Gochartów. Lista 402 identyfikatorów zestawu sond Affymetrix została opatrzona adnotacją z przypisanymi do proteomu klasyfikacjami funkcjonalnymi dostarczonymi przez LocusLink., Procent pokrycia reprezentuje liczbę genów spośród 402 które zostały adnotowane na poziomie specyficzności terminowej w procesie biologicznym, funkcji Molekularnej i klasyfikacji komponentów komórkowych. Zasięg procentowy maleje wraz ze wzrostem specyficzności terminowej.
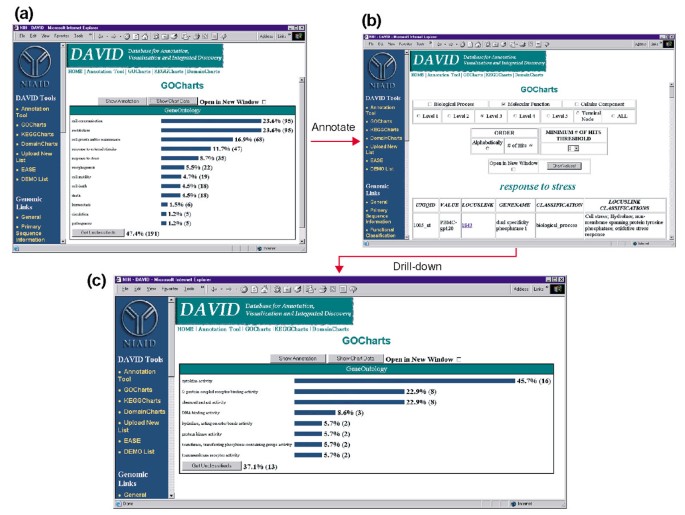
ouput gocharts. (a) wykres słupkowy pokazujący rozkład genów wyrażonych różnie między procesami biologicznymi ontologii genów (GO)., Parametry zostały ustawione na poziom 2, próg trafienia 5, a wynik został posortowany według liczby trafień. Niebieskie paski są połączone z dodatkowymi danymi adnotacji pokazanymi w lit. b). Wybranie niebieskiego paska w (a) odpowiadającego „odpowiedź na stres” otwiera tabelę HTML pokazującą LocusLink, nazwę genu, aktualną klasyfikację i inne dane klasyfikacyjne dla genów w tej kategorii. (c) ten podzbiór genów zaangażowanych w „Odpowiedź na stres” był dalej scharakteryzowany przez wybór Go funkcji molekularnej, GO poziom 3, próg hit 2, i posortowane przez hit count., Wybranie przycisku „wartości wykresu” tworzy nowy histogram ujawniający, że 16 z 35 genów odpowiedzi stresowej koduje białka posiadające aktywność cytokin.,
ponieważ HIV-1 ma duży wpływ na funkcję komórek układu odpornościowego i ich zdolność do przeprowadzania reakcji stresowych, wybraliśmy Pasek histogramu reprezentujący liczbę genów zaangażowanych w reakcję stresową, który otwiera tabelę HTML zawierającą identyfikator Affymetrix, numer LocusLink, nazwę genu, aktualną klasyfikację i inne klasyfikacje dla wszystkich 35 genów (ryc. 4b)., Teraz, gdy zredukowaliśmy naszą listę genów do tych genów zaangażowanych w odpowiedzi na stres, scharakteryzowaliśmy ten podzbiór, powtarzając procedurę Gochartsa dostępną na górze tabeli HTML odpowiedzi na stres. Wybierając funkcję molekularną, poziom 3 tworzy nowy histogram, który szybko ujawnia, że prawie połowa (16/35) genów odpowiedzi stresowej posiada aktywność cytokin (ryc. 4c)., Rzeczywiście, Wykazano, że cytokiny odgrywają ważną rolę w cyklu życia HIV-1, a uzyskane wyniki sugerują, że leczenie PBMC białkami otoczki HIV-1 znacząco moduluje transkrypcję licznych genów cytokin. Wydajność, z jaką GoCharts systematycznie podsumowywał ten duży zbiór danych za pomocą graficznych wizualizacji, pozostając jednocześnie połączonym z podstawowymi danymi i zewnętrznymi zasobami, znacznie poprawiła proces wykrywania.,
KeggCharts
rysunek 5a przedstawia wynik KeggCharts z histogramem pokazującym rozkład genów wyrażonych różnie wśród szlaków biochemicznych. Wykres pokazuje, że szlak apoptozy KEGG obejmuje pięć genów indukowanych przez gp120 HIV-1. Wybranie nazwy szlaku otwiera odpowiednią mapę szlaku biochemicznego KEGG i zaznacza na Czerwono różnie wyrażone geny funkcjonujące w tym szlaku (rysunek 5b). W tym ujęciu geny są dodatkowo powiązane z dodatkowymi adnotacjami dostępnymi za pośrednictwem systemu pobierania DBGET KEGG ., Zauważ, że tylko cztery geny w szlaku apoptozy KEGG są zaznaczone na czerwono, podczas gdy narzędzie Keggcharts mapowało pięć zestawów sondy Affymetrix do szlaku apoptozy. Różnica ta wynika z faktu, że dwa sondy Affymetrix celują w ten sam gen „TNF-alfa”.
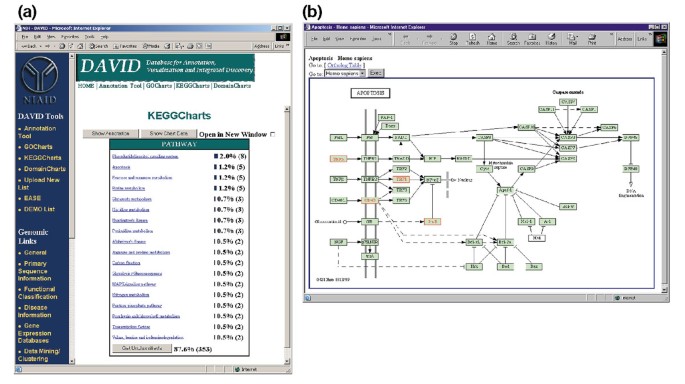
zestaw Keggchartów. (a) Wykres wizualizacji pokazujący rozkład 402 genów wśród szlaków biochemicznych KEGG. Próg trafienia został ustawiony na trzy, a wynik został posortowany według liczby trafień., Duża liczba niekategoryzowanych identyfikatorów wynika z faktu, że KEGG jest biochemicznie skoncentrowany, a tym samym zapewnia niski zasięg list genów. Podobnie jak wyjście Gochartów, niebieskie paski reprezentują liczbę genów w każdej ścieżce. Wybranie niebieskiego paska otwiera tabelę HTML pokazującą LocusLink, nazwę genu, aktualną klasyfikację i inne dane klasyfikacyjne dla genów w tej ścieżce (dane nie są pokazane)., b) szlak biochemiczny KEGG, który pojawia się po wybraniu nazwy szlaku „apoptosis” w lit. a), przedstawia cztery geny o różnej ekspresji w obrębie szlaku apoptozy, podkreślając je jasnozielonym i czerwonym kolorem. Fakt, że szlak KEGG podkreśla tylko cztery geny, podczas gdy keggchart mapuje pięć zestawów sondy Affymetrix do szlaku apoptozy wynika z faktu, że dwie sondy ustawiają cel tego samego genu „TNF-alfa”.,
DomainCharts
DomainCharts są operacyjnie podobne do KeggCharts i GoCharts, z wyjątkiem tego, że wyniki wizualnie obrazują rozkład genów wśród domen białka PFAM (rysunek 6a). Histogram DomainCharts identyfikuje 16 genów z domenami kinazy (pkinase), prawdopodobnie odzwierciedlając wpływ HIV-1 gp120 na maszynerię transdukcji sygnału. Wykres identyfikuje również sześć genów z domenami interleukiny-8 (IL-8), które stanowią wysoce zachowany motyw wśród cytokin odpowiedzi stresowej., Wybranie nazwy domeny ” IL8 ” otwiera stronę Conserved Domain Database (CDD) odpowiadającą tej domenie PFAM (rysunek 6b). Ta strona zawiera szczegółowe sekwencje, strukturę i funkcjonalne informacje o domenie IL-8 i białkach, które ją zawierają.
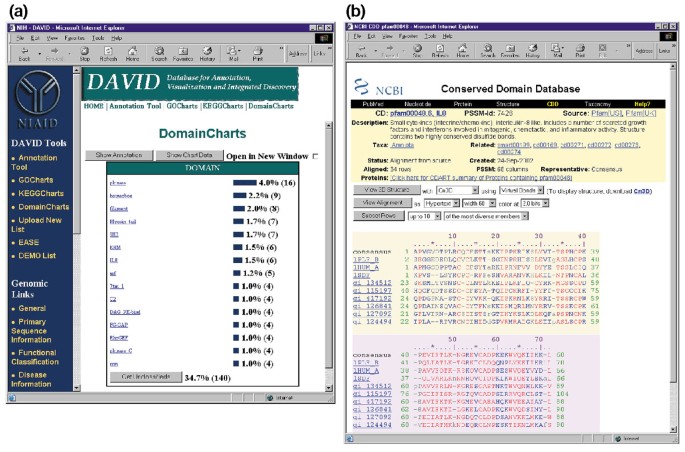
Ouput DomainCharts. (a) Wykres wizualizacji pokazujący rozkład 402 genów wśród domen białkowych. Parametry zostały ustawione na minimalny próg trafień wynoszący cztery, a wyniki zostały posortowane według liczby trafień., Podobnie jak w przypadku Gochartów i Keggchartów, niebieskie paski reprezentują liczbę genów zawierających daną domenę. Wybranie niebieskiego paska otwiera tabelę HTML pokazującą LocusLink, nazwę genu, aktualną klasyfikację i inne dane klasyfikacyjne dla genów w tej ścieżce (dane nie są pokazane)., (b) wybranie nazwy domeny” IL8 ” w (A), która zawiera sześć różnie wyrażonych genów, przenosi użytkownika na nową stronę zawierającą dane wyjściowe z bazy danych Conserved Domain Database (CDD) NCBI, która dostarcza szczegółowych informacji o domenie IL-8, w tym informacji strukturalnych, wielu wyrównań sekwencji i informacji opisowych o domenie i białkach, które ją posiadają.