wprowadzenie
śmiertelność prosiąt jest jedną z głównych cech selekcji w hodowli świń i zależy od lochy, prosiąt i środowiska. Dlatego śmiertelność prosiąt jest złożonym fenotypem i zależy od zdolności lochy do wychowania potomstwa, ale jest również funkcją wagi urodzeniowej, zarządzania i selekcji (Knol et al., 2002)., Jednak również monogenne wady recesywne przyczyniają się do śmiertelności prosiąt, chociaż w przeszłości odnotowano tylko kilka przykładów (Murgiano et al., 2012; Matika et al., 2019). Nawet w tych przypadkach, gdy efekt mutacji jest poważny, skuteczny wybór przeciwko takiej mutacji jest utrudniony przez niską częstotliwość. W wielu poważnych wadach zygota umierają bardzo wcześnie w ciąży, nie pozostawiając żadnych śladów innych niż brak homozygotów w populacji na ogół(Derks et al., 2019).,
efekty chowu wsobnego w komercyjnych populacjach świń są zwykle kontrolowane przez selektywną hodowlę w celu zmniejszenia śmiertelności prosiąt poprzez poprawę zarówno zdolności matecznych, jak i żywotności prosiąt (Olijslagers, 2018). Jednak warianty leżące u podstaw recesywnych defektów monogennych nie są dobrze uchwycone w wartościach hodowlanych i potencjalnie dryfują do wyższych częstotliwości w wyniku intensywnej selekcji(Georges et al., 2019). Ponadto warianty te mogą być również utrzymane w wyniku równoważenia selekcji dla skorelowanego pozytywnego efektu w stanie heterozygotycznym (Derks et al., 2018).,
wady recesywne tylko nieznacznie przyczyniają się do ogólnej śmiertelności prosiąt (Alonso-Spilsbury et al., 2007). Niemniej jednak warianty wpływające na śmiertelność prosiąt mają ogromne znaczenie, ponieważ warianty te bezpośrednio wpływają na produkcję i dobrostan zwierząt (Baxter et al., 2013; Rutherford et al., 2013). Jednak w zarządzaniu populacją zwierząt występowanie wad niskiej częstotliwości jest zwykle słabo udokumentowane (często używa się bardzo ogólnych terminów), a zespoły są często rozpoznawane dopiero po osiągnięciu wysokiej częstotliwości., Jest to szczególnie istotne w przypadku zespołów, które nie prowadzą do bardzo wyraźnych fenotypów. W związku z tym nawet w populacjach hodowli komercyjnej niewiele można śledzić na określonych zespołach, a skuteczne dobranie do konkretnych zespołów o niskiej częstotliwości wymaga zatem nowych podejść.
w niniejszej pracy opisujemy odkrycie wysoce wyniszczającego zespołu w komercyjnej populacji świń poprzez badanie oparte na połączonych tablicach SNP o średniej gęstości i sekwencjonowaniu całego genomu (WGS)., Badanie doprowadziło do identyfikacji delecji 16-bp frameshift w genie SPTBN4, z przewidywanymi wyraźnymi konsekwencjami fenotypowymi w homozygotach. Częstotliwość nośna wynosi około 9% w badanej populacji, wpływając na około 0,81% populacji. Częstość była na tyle niska, że nie była znana, aby mieć podłoże genetyczne, a nawet w ogóle nie była rozpoznawana jako konkretny zespół. Po przeprowadzeniu badania jedna ciężarna locha została zidentyfikowana jako spłodzona przez dzika przewoźnika., Chore prosięta cierpią na miopatię i nie są w stanie chodzić, co zwykle skutkuje śmiercią w ciągu kilku godzin po urodzeniu, całkowicie zgodnie z przewidywaną patologią w porównaniu do podobnych przypadków u ludzi i myszy.
materiał i metody
Zwierzęta, genotypy i obróbka wstępna
zbiór danych składa się z 31 839 zwierząt z syntetycznej linii dzika o dużym białym tle. Linia jest utrzymywana i hodowana w gospodarstwach Topigs Norsvin Nucleus, przede wszystkim wybierając cechy produkcyjne i zdrowotne., Zwierzęta zostały genotypowane na chipie Illumina GeneSeek custom 50K SNP (Lincoln, NE, USA). Zwierzęta z częstością występowania brakujących genotypów > 0,15 zostały usunięte. Odrzuciliśmy markery, które nie spełniały następujących kryteriów filtrowania: minimalna częstotliwość wywołania 0,85, mniejsza częstotliwość alleli >0,01, oraz dokładna wartość P w proporcjach Hardy ' ego-Weinberga poniżej P< 10-12. Co więcej, markery o nieznanej lokalizacji na budowie genomu Sscrofa11.1 zostały odrzucone, pozostawiając 41 573 markery po filtrowaniu., Wszystkie kroki zostały wykonane w Plink v1.90b3. 30 (Purcell et al., 2007).
haplotype Phasing i identyfikacja Haplotype Ssc6
wykonaliśmy haplotype phasing i przypisanie brakujących miejsc w Beagle5.0 z parametrem efektywnej wielkości populacji ustawiony na 100, inne ustawienia były domyślne (Browning et al., 2018). Oczekiwane homozygoty oszacowano na podstawie częstości haplotypów, stosując zasadę Hardy ' ego-Weinberga. Dokładny test dwumianowy został zastosowany w celu sprawdzenia liczby obserwowanych homozygot z liczbą oczekiwanych homozygot., Haplotyp został uznany za znacznie uszczuplony, jeśli P < 5 × 10-3.
efekty fenotypowe związane z Haplotypem SSC6
zbadaliśmy haplotyp SSC6 pod kątem rekordów całkowitej liczby urodzonych, liczby martwych urodzonych, zmumifikowanych prosiąt, przeżycia porodowego i przeżycia laktacji (przeżycie do około 21 dni) w sumie 9666 miotów. Wymieniliśmy te fenotypy dla wszystkich zidentyfikowanych miotów CxC i CxN. Użyliśmy testu t Welcha, aby ocenić, czy fenotypy pochodzące z miotów CxC różnią się znacząco od miotów CxN. A wartość p < 0.,05 został uznany za znaczący.
Analiza sekwencjonowania całego genomu i identyfikacja wariantu kandydata
zbiór danych składa się z 71 osobników sekwencjonowanych całego genomu z badanej populacji. Wszystkie 71 próbek były również obecne w naszym zestawie danych 31 839 zwierząt genotypowanych na 50k. 71 próbek ma całkowitą objętość 1,93 Tbp (TERA base pairs) z 14,16 miliardów 150-bp sparowanych odczytów końcowych (tabela S3). Próbki zostały zsekwencjonowane na Illumina HiSeq 2000. Dopasowaliśmy sekwencje do genomu Sscrofa11.1 używając BWA-mem w wersji 0.7.,15 (Li i Durbin, 2009) ze średnią mapowalnością 98,9% i pokryciem próbki w zakresie od 8,8 do 14,8 X (Średnia 10,9 X). Samblaster został użyty do usuwania duplikatów PCR (Faust and Hall, 2014). Samtools był używany do sortowania, scalania i indeksowania plików bam (Li et al., 2009). Mapowanie i statystyki jakości zostały wygenerowane za pomocą Qualimap (Okonechnikov et al., 2016). Wywołanie wariantu zostało wykonane z Freebayes v1.1.0 z następującymi ustawieniami: –min-base-quality 10-min-alternate-fraction 0.2-haplotype-length 0-min-alternate-count 2 (Garrison and Marth, 2012)., Warianty z wynikiem jakości Phred < 20 zostały odrzucone (Li et al., 2009). Warianty zostały opatrzone adnotacją za pomocą Ensembl variant effect predictor (VEP, release 96) (Mclaren et al., 2016). Wpływ wariantów missense przewidywano za pomocą sortowania nietolerancji od tolerancyjnego (SIFT) (Kumar et al., 2009). Analiza LD została przeprowadzona przy użyciu Plink v1.90b3. 30 (Purcell et al., 2007) z następującymi ustawieniami –Chr-set 18, –r2, LD-window-r2 0.8.
wyrównanie białka SPTBN4
wyrównanie białka między rodzajem dzikim a zmutowanym białkiem przeprowadzono za pomocą ClustalO (Madeira et al.,, 2019) i wizualizowane za pomocą ESPript 3 (Robert and Gouet, 2014). Dalsza wizualizacja i Walidacja została przeprowadzona przy użyciu jbrowse genome viewer w wersji 1.12.1 (Skinner et al., 2009).
walidację przyczynowej delecji 16 bp SPTBN4
PCR przeprowadzono przy użyciu 60 ng genomowego DNA, z 0,4 µm każdego podkładu, 1,8 mM MgCl2 i 25 jednostek / ml polimerazy DNA ONETAQ® (Onetaq® 2x Master Mix with Standard Buffer, New England Biolabs) w buforze PCR producenta w końcowej objętości 12 µl., Początkowo denaturacja przez 1 min w temperaturze 95°C następowała przez 35 cykli 95°C przez 30 s, 55°C przez 45 s, 72°C 90 s, a następnie 5 minut przedłużeniem 72°C. primerami PCR dla SPTBN4 są TCAAGGGTGCAGCTCTTTC do przodu i ggtaggaagctcgaagtgg do tyłu. Przedni podkład był oznaczony barwnikiem 6-FAM, aby wytworzyć fluorescencyjnie oznakowany produkt PCR wykrywalny na sekwenatorze DNA ABI 3730 (Applied Biosystems). Rozmiary fragmentów określono za pomocą oprogramowania GeneMapper 5 firmy ABI.,
badanie histopatologiczne
dwa chore prosięta w wieku poniżej 1 tygodnia zostały wysłane do działu patologii Royal Animal Health (Deventer) w celu zbadania. Makroskopowo wszystkie obserwacje mieściły się w granicach normy. Pobrano próbki mięśni szkieletowych kończyny przedniej, grzbietu i tylnej nogi obu zwierząt w celu rutynowego barwienia H&barwienia E i PTAH. Tkankę mięśniową przechowywano w oddzielnych słoikach i utrwalano w roztworze formaldehydu 4%, buforowanym (=roztwór formaliny 10%, buforowany)., Następnie tkanka została osadzona w parafinie i pocięta na 2 µm zgodnie ze standardową procedurą operacyjną (SOP RAH). Następnie szkiełka były odparafinowane i rutynowo barwione na hematoksylinę i eozynę (H& E) w automatycznej maszynie kolorowej. Jednocześnie przygotowano dodatkowe szkiełka 2 µm tkanki mięśniowej, a także pozytywną szkiełkę kontrolną tkanki mięśniowej do ręcznego barwienia „hematoksyliną kwasu fosfotungstowego”, w skrócie PTAH. Barwienie to jest korzystne dla wykazania poprzecznego prążkowania mięśni szkieletowych.,
wartości hodowlane i analiza Asocjacji
w tym badaniu oceniliśmy 63 cechy wykorzystywane w programie hodowlanym. Deregressed estimated breeding values (DEBV) były używane jako zmienna odpowiedzi dla każdej badanej cechy. Szacowana wartość hodowlana (EBV) wszystkich ocenianych cech została poddana deregresji przy użyciu metodologii opisanej przez Garrick et al. (2009). EBV każdego zwierzęcia uzyskano na podstawie rutynowej oceny genetycznej w komercyjnym programie hodowlanym (Topigs Norsvin) z wykorzystaniem modelu zwierzęcego., Wiarygodność na zwierzę w celu deregresji została wyodrębniona z oceny genetycznej opartej na metodologii Tier I Meyer (2004). Dziedziczność użyta do deregresji została również wyodrębniona z rutynowej oceny genetycznej. Wreszcie, współczynniki ważenia oparte na szacowanej wiarygodności DEBV zostały również oszacowane zgodnie z Garrick et al. (2009) używając wartości 0,5 dla skalara c. w celu zapewnienia jakości DEBV, tylko zwierzęta o współczynnikach wagowych większych niż zero i wiarygodności DEBV większej niż 0.,W analizach Stowarzyszenia wykorzystano 20. Wiarygodność DEBV została również uzyskana według Garrick et al. (2009).
analizy asocjacyjne przeprowadzono przy użyciu oprogramowania ASREML (Gilmour et al.,, 2009) stosując następujący liniowy mieszany model zwierzęcy:
gdzie DEBVij jest obserwowanym DEBV dla zwierzęcia j, W jest współczynnikiem wagowym dla resztkowego, µ jest ogólną średnią DEBV w populacji, Ri jest nośnikiem (liczbą szkodliwego allelu) 4 mutacji i, aj jest addytywnym efektem genetycznym szacowanym przy użyciu matrycy średniej relacji opartej na rodowodzie, a śladowy wpływ błąd. Skojarzenia z a-log10 (wartość P) większa niż pięć zostały uznane za znaczące.
wyniki
A 1.,5 Mb segmentu na chromosomie 6 wpływa na przeżywalność laktacji u świń
przeanalizowaliśmy 31,638 zwierząt z jednej linii dzika czystej krwi (linia syntetyczna z dużym białym tle), genotypowane na świni 50K SNP chip (sscrofa11. 1 budować) (Warr et al., 2019). Analiza wykazała segment 1,5 Mb na chromosomie 6 (SSC6:48,75-50,25) wykazujący deficyt homozygotyzmu związany ze zmniejszonym przeżywaniem laktacji (tabele 1 i 2). Haplotyp segreguje się z umiarkowaną częstością alleli wynoszącą 4,5% (9,0% częstotliwości nośnej) w badanej populacji., Częstotliwość haplotypów wahała się w ciągu ostatniej dekady, ale zmniejszyła się w ciągu ostatnich 3 lat (rysunek S1). Sprawdziliśmy, czy częstotliwość była napędzana przez heterozygotyczny efekt przewagi. Stwierdzono jednak głównie negatywne skojarzenia z ważnymi cechami selekcji, z wyjątkiem głębokości schabu i długości ciąży (Tabela 3), co sugeruje, że częstotliwość jest wyłącznie wynikiem dryfu genetycznego.

tabela 1 ssc6,

tabela 2 mioty po nosicielach wykazują 24% spadek przeżywalności laktacji w porównaniu do miotów po nosicielach innych niż nosicielskie. Znaczące wyniki zaznaczono pogrubioną czcionką.

Tabela 3 Cechy istotnie związane z heterozygotycznymi nośnikami delecji SPTBN4.
52 mioty typu carrier-by-carrier (CxC) nie wykazują znaczącego zmniejszenia całkowitej liczby urodzonych lub żywych zwierząt., Jednak przeżywalność laktacji jest zmniejszona o około 24% w miotach CxC w porównaniu do krycia typu carrier-by-noncarrier (CxN), co wskazuje, że homozygotyczne prosięta umierają w okresie laktacji (Tabela 2). Następnie zbadaliśmy uwagi dotyczące czasu i przyczyny śmiertelności miotów CxC. To ujawniło, że większość prosiąt, które zmarły w ciągu pierwszych 24 godzin po urodzeniu. Większość tych prosiąt była w większości opisywana przez rolników jako ” słabe prosiąt po urodzeniu.,”
Analiza sekwencjonowania całego genomu ujawnia delecję 16-bp Frameshift w SPTBN4 jako prawdopodobny wariant przyczynowy
aby zidentyfikować mutację przyczynową, zbadaliśmy dane sekwencji całego genomu z 71 zwierząt z badanej populacji i zidentyfikowaliśmy pięć zwierząt nośnych. Analiza powiązania disequilibrium (LD) wykazała 267 wariantów SNP i indel w wysokim LD (R2 > 0.8) z haplotypem SSC6 (tabela S1), większość w idealnym LD (247 wariantów)., Tylko pięć wariantów potencjalnie wpływa na sekwencję Kodowania (trzy missense, jeden frameshift, jeden Splice-acceptor). Przewiduje się, że trzy warianty missense będą tolerowane przez SIFT (wynik > 0.18, tabela S1), podczas gdy wariant splice-acceptor wpływa na gen kodujący peptyd 28 bp o nieznanej funkcji, mało prawdopodobne, aby był przyczynowy. Jednak jeden wariant w całości LD (r2 = 1) z haplotypem miał duży wpływ; delecja 16-bp frameshift w eksonie 26 genu SPTBN4 (6: g. 48801280delgacggtgtacgccggt) (ryc. 1A, B)., Delecja frameshift (ENSSSCP00000031537:P. Arg1902fs) wprowadza 30 nowych aminokwasów i przedwczesny kodon stop, wytwarzając upośledzone i okrojone białko spectrin beta non-erytrocytic 4 (SPTBN4). Mutantom brakuje końcowych 662 aminokwasów białka typu dzikiego (rysunek 1C), w tym domeny homologii pleckstriny (PH) wymaganej do transportu białka do błon (Wang et al., 2018). Białko SPTBN4 należy do białek beta-spectryny i jest aktyną, która łączy błonę komórkową z cytoszkieletem aktyny., Mutacje SPTBN4 zakłócają maszynerię cytoszkieletową kontrolującą prawidłową lokalizację kanałów jonowych w mielinizowanych nerwach powodujących neuropatie ruchowe (Parkinson et al., 2001; Wang et al., 2018).

Rysunek 1 (A) Model genu SPTBN4. Położenie 26. egzonu jest zaznaczone na Czerwono. B) Ilustracja skreślenia 16-bp. Rysunek pokazuje dziki typ i zmutowany exon. C) wyrównanie sekwencji białka sptbn4 mutanta (Mt) i typu dzikiego (WT). Mutacja indukuje 30 nowych aminokwasów i przedwczesny kodon stop.,
genotypowanie pięciu miotów CxC potwierdza delecję SPTBN4 jako prawdopodobnego winowajcę
genotypowaliśmy pięć miotów CxC dla delecji 16-bp, które miały co najmniej dwa prosięta (zakres 2-6), które zmarły w ciągu pierwszych 48 godzin po urodzeniu. W pięciu miotach wyprodukowano 53 prosięta, z których 19 było homozygotycznych dla delecji 16 bp (Tabela 4). Wszystkie 19 homozygotycznych prosiąt zmarło w ciągu 48 godzin od urodzenia (18 w ciągu 24 godzin). Z 34 pozostałych prosiąt (8 dzikich i 26 nosicieli) tylko 1 zmarło w ciągu 48 godzin, prawdopodobnie z powodu innych (środowiskowych) czynników.,

Tabela 4 genotypowanie prawdopodobnej przyczynowej delecji frameshift 16-bp SPTBN4 w pięciu miotach po nosicielu. Pogrubioną czcionką zaznaczono sumę na klasę genotypu.
prosięta homozygotyczne z powodu delecji SPTBN4 cierpią na miopatię i paraliż kończyn tylnych
monitorowaliśmy jeden niedawny miot CxC (data porodu: 28 kwietnia 2019 r.), w którym urodziło się sześć zdrowych, dwa dotknięte chorobą (próbki: 9912, 9916) (ryc., Potwierdziliśmy homozygotyczny status delecji SPTBN4 dla dwóch dotkniętych nimi prosiąt (tabela S2). Ponadto wśród zdrowych osobników zaobserwowano cztery heterozygotyczne nosiciele i dwa homozygotyczne prosięta typu dzikiego. Jedno z martwych prosiąt (próbka: 9921) również było homozygotyczne dla delecji, podczas gdy pozostałe dwa były heterozygotyczne. Dotknięte prosięta cierpią z powodu skrajnego osłabienia mięśni (ryc. 2b, C), paraliżu kończyn tylnych i drżenia (wideo S1). Stąd prosięta nie były w stanie chodzić ani pić.,
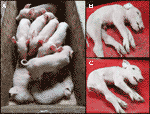
Rysunek 2 (a) dwa chore prosięta (żywe) wraz z sześcioma zdrowymi młodymi. Prosięta pochodzą z jednego porodu godowego CxC, odłowionego 28 kwietnia 2019 roku. B) dotknięty samiec prosiaka 9912. C) dotknięta samica Prosiaczka 9916.
chore prosięta nie mają poprzecznych prążków w mięśniach szkieletowych kończyn grzbietowych i tylnych
badanie histopatologiczne wykazało rozproszone zwyrodnienie włókien mięśniowych u obu prosiąt oraz martwicę ogniskową i zapalenie naczyń w mięśniu grzbietowym u jednego z prosiąt (ID = 9912)., Ponadto barwienie hematoksylin kwasem fosfotungstowym (PTAH) wykazuje rozbieżne zabarwienie włókien mięśni szkieletowych, co wskazuje na zmniejszenie poprzecznego prążkowania, szczególnie w mięśniach grzbietowych i tylnych nóg dotkniętych zwierząt (ryc. 3b), podczas gdy przednie nogi wydają się nienaruszone (ryc. 3A). Na zmniejszenie poprzecznego prążkowania wskazuje nieprawidłowe zabarwienie i ogólna utrata objętości włókien mięśniowych (ryc. 3b). Histopatologicznie obserwowane zmiany w kończynach tylnych i mięśniach grzbietowych wskazują na dystrofię mięśniową.,
Rysunek 3 (a) przekrój mięśni szkieletowych przednich nóg. Czarna strzałka wskazuje normalne zabarwienie (ciemne) włókien mięśniowych wskazując na obecność poprzecznych prążków. Bar PTAH = 50 µm. B) przekrojowy widok mięśnia szkieletowego z tylnej nogi. Czarna strzałka wskazuje nieprawidłowe zabarwienie (różowe) włókien mięśniowych wskazując na brak poprzecznych prążków. Żółta strzałka wskazuje normalne zabarwienie i obecność poprzecznych prążków. Bar PTAH = 50 µm.,
dyskusja
w niniejszej pracy zgłaszamy nową wadę wrodzoną powodującą śmiertelność prosiąt prawdopodobnie z powodu delecji 16 bp w genie SPTBN4. Prosięta cierpią na skrajne osłabienie mięśni (miopatię) i umierają w ciągu kilku godzin po urodzeniu. Oczekuje się, że delecja spowoduje całkowitą utratę funkcji spektryny beta, nie erytrocytycznego białka 4. SPTBN4 należy do rodziny genów spectrin i jest wymagany do klastrowania kanałów jonowych w węzłach Ranviera, wpływając na potencjał czynnościowy (Devaux, 2010)., Mutacje zaburzają cytoszkieletową maszynerię, która kontroluje prawidłową lokalizację kanałów jonowych i funkcję domen aksonalnych głównie w początkowych segmentach aksonu (AIS) i węzłach Ranviera (Wang et al., 2018). Dokładniej, dotknięta domena terminala C sptbn4 ma kluczowe znaczenie dla handlu kanałami KCNQ2 i pobudliwości w węzłach Ranviera (Devaux, 2010).
kolejne badania kontrolne zidentyfikowały przypadki ludzi i myszy, które wskazywały, że wynikający z nich zespół prawdopodobnie nie okaże się natychmiast śmiertelny, ale raczej spowodowałby ciężką miopatię., Na podstawie danych o genotypie SNP o średniej gęstości, dostępnych dla wszystkich zwierząt w populacji hodowlanej (N = 31 839), można było zidentyfikować nosicieli. Wśród tych nosicieli była locha, która była w połowie ciąży w momencie identyfikacji, spłonięta przez dzika, który był również nosicielem. Gospodarstwo hodowlane zostało powiadomione o udokumentowaniu miotu po urodzeniu. Obserwowany fenotyp prosiąt dotkniętych chorobą (miopatia, porażenie kończyn tylnych, drżenia) był całkowicie zgodny z tym, co zaobserwowano u ludzi z homozygotyczną utratą funkcji lub złożonymi heterozygotycznymi mutacjami w genie SPTBN4 (OMIM: 606214)., Dwóch pacjentów u ludzi ma mutacje utraty funkcji w obrębie domeny PH (Wang et al., 2018), potwierdzając, że utrata domeny PH u świń prawdopodobnie doprowadziłaby do całkowitej utraty funkcji białka SPTBN4. U ludzi podobne mutacje prowadzą do ciężkiej wrodzonej miopatii spowodowanej brakiem włókien mięśniowych typu i, neuropatią i głuchotą (Knierim et al., 2017; Wang et al., 2018). Wang et al. (2018) zaobserwowano również neuropatię aksonalną motoryczną u kilku pacjentów charakteryzujących się wrodzoną hipotonią, głębokim osłabieniem i utratą głębokich odruchów ścięgnistych we wczesnym dzieciństwie., Ponadto biopsje nerwów wykazały zredukowane węzłowe kanały Na+ i brak węzłowych kanałów KCNQ2 K+, ujawniając patologię molekularną powodującą dysfunkcję układu nerwowego. W związku z tym wnioskujemy, że ten wariant zmiany ramki jest prawdopodobną mutacją przyczynową prowadzącą do obserwowanego fenotypu i zubożenia homozygotycznego genotypu w populacji. Przyszłe badania mogłyby skupić się na dokonywaniu in vivo knockoutu genu SPTBN4 u świń, aby dokładniej zbadać zespół i związany z nim fenotyp.,
nie zaobserwowaliśmy degeneracji włókien mięśniowych przednich nóg, podczas gdy włókna mięśniowe grzbietowe i tylne były wyraźnie dotknięte. Ta obserwacja może częściowo wyjaśnić paraliż kończyny tylnej, podczas gdy przednie nogi nie są dotknięte. Rozbieżność między włóknami mięśniowymi przednich i tylnych nóg została również opisana u drżących myszy, u których mutacje utraty funkcji SPTBN4 powodują neuropatię ruchową, paraliż kończyn tylnych, drżenia i głuchotę centralną (Parkinson et al., 2001; Komada i Soriano, 2002). Parkinson i in., (2001) opisz zmniejszone prędkości przewodzenia nerwów w nerwach kulszowych myszy z drżącymi allelami powodującymi obwodową neuropatię kończyn tylnych. Ekspresja SPTBN4 u myszy jest ograniczona do mózgu, rdzenia kręgowego i nerwów kulszowych i nie obserwuje się jej w mięśniach szkieletowych, więc choroba ta jest przede wszystkim defektem neuronalnym. Ogólnie nie wiadomo, który mechanizm powoduje brak objawów w kończynach przednich. Ten „naturalny nokaut” u świń może być użytecznym zasobem do badania ludzkiej choroby, ponieważ świnie są zwykle lepszym modelem do badania ludzkiej choroby w porównaniu do gatunków gryzoni., Co więcej, konsekwencje utraty funkcji SPTBN4 można zbadać bardziej szczegółowo.
efektywna wielkość populacji (Ne) badanej rasy szacuje się na około 100 (Hidalgo et al., 2016). W hodowli zwierząt niski poziom Ne zwiększa ryzyko przypadkowego wzrostu częstości alleli. Co więcej, poprzednie badania wykazały, że recesywne allele śmiertelne mogą być napędzane przez korzystne efekty w heterozygotach (Derks et al., 2018; Matika et al., 2019). Matika i in.,, 2019 stwierdzono recesywną mutację stop-uzyskaną w genie MSTN związaną z znacznym wzrostem głębokości mięśni u heterozygotów. Jednak nie znajdujemy dowodów na jakąkolwiek heterozygotyczną przewagę w naszym badaniu. Dzięki obecnym technikom genomicznym możemy teraz zidentyfikować szkodliwe allele dryfujące do wyższych częstotliwości i dokładnie monitorować pojawienie się nowych szkodliwych alleli, umożliwiając bardziej skuteczne oczyszczanie. Co więcej, wynik tego typu badań znacznie poprawi świadomość” ukrytych ” wad genetycznych zarówno na poziomie hodowcy, jak i rolnika., Bez żadnych wcześniejszych informacji rzadkie wady wrodzone są często rejestrowane jako ” słaby Prosiaczek.”I bez dalszego rozróżniania konkretnych zespołów, dalsze działanie nie jest możliwe. W większości przypadków nie wiadomo, czy istnieje podłoże genetyczne lub czy mogą wystąpić inne zakłócające skutki. Z wcześniejszych informacji genomicznych, zespół może być zidentyfikowany, w porównaniu do innych przypadków, i nośniki zidentyfikowane, co prowadzi do zaskarżalnych informacji.
śmiertelność prosiąt ma duże znaczenie gospodarcze i dobrostanu zwierząt., Dlatego odkrycie mutacji SPTBN4 doprowadziło do natychmiastowego wdrożenia w programie hodowlanym, aby zminimalizować częstotliwość krycia po nosicielu. Pozwala to uniknąć narodzin osób dotkniętych chorobą, poprawiając w ten sposób dobrostan zwierząt i zmniejszając straty ekonomiczne.
wnioski
w niniejszym badaniu zgłaszamy nową wadę wrodzoną, prawdopodobnie spowodowaną recesywną delecją w genie SPTBN4 u świń. Wyniki są poparte uderzającymi podobieństwami do fenotypów związanych z zespołem SPTBN4 u ludzi i myszy., Badanie pozwala monitorować i usuwać szkodliwe allele z populacji. Można zapobiegać krzyżom między przewoźnikami, wykluczając osoby dotknięte chorobą, zmniejszając tym samym straty ekonomiczne i poprawiając dobrostan zwierząt. Wreszcie ,te” naturalne nokauty ” uzyskane w przemyśle hodowlanym mogą stanowić model dla ludzkiej choroby i zwiększyć nasze zrozumienie funkcji genów w kladzie ssaków i zapewnić potencjalny model dla ludzkiej choroby.,
Oświadczenie o dostępności danych
Oświadczenie o etyce
przegląd etyczny i zatwierdzenie nie były wymagane w przypadku badania na zwierzętach, ponieważ dane wykorzystane w tym badaniu zostały uzyskane w ramach rutynowego gromadzenia danych z programów hodowlanych Topigs Norsvin, a NIE specjalnie na potrzeby tego projektu. Dlatego też zatwierdzenie komisji etyki nie było obowiązkowe. Pobieranie próbek i rejestrowanie danych przeprowadzono ściśle zgodnie z holenderską ustawą o ochronie i dobrostanie zwierząt (Gezondheids – en welzijnswet voor dieren)., Pisemną świadomą zgodę uzyskano od właścicieli na udział ich zwierząt w tym badaniu.
wkład autora
BH była odpowiedzialna za ogólną organizację i komunikację z Topigs Norsvin i rolnikami. MD I ML przeprowadziły analizę danych. BD i KL wykonały prace laboratoryjne. SG-V przeprowadził analizę patologiczną. MD napisał manuskrypt. H-JM, MG, BH, SG-V, BD, KL I ML dostarczyły użytecznych uwag i sugestii oraz pomogły w opracowaniu rękopisu. Dane fenotypowe analizowano w ml., Wszyscy autorzy przeczytali i zatwierdzili ostateczny manuskrypt.
finansowanie
badanie zostało sfinansowane przez partnerstwo stw-Breed4Food, numer projektu 14283: od sekwencji do fenotypu: wykrywanie szkodliwej zmienności przez przewidywanie funkcjonalności. Badanie to było wspierane finansowo przez NWO-TTW oraz partnerów Breed4Food Cobb Europe, CRV, Hendrix Genetics i Topigs Norsvin. Ponadto badanie to było wspierane przez projekt IMAGE (Horizon 2020, nr 677353). Fundatorzy nie mieli żadnej roli w projektowaniu studiów, zbieraniu i analizie danych, decyzji o publikacji lub przygotowaniu rękopisu., Wykorzystanie klastra HPC było możliwe dzięki CATAgroFood (Shared Research Facilities Wageningen ur).
Disclaimer
dane wykorzystane w tym badaniu zostały uzyskane w ramach rutynowego zbierania danych z programów hodowli Topigs Norsvin, a NIE specjalnie na potrzeby tego projektu. Dlatego też zatwierdzenie komisji etyki nie było obowiązkowe. Pobieranie próbek i rejestrowanie danych przeprowadzono ściśle zgodnie z holenderską ustawą o ochronie i dobrostanie zwierząt (Gezondheids – en welzijnswet voor dieren).,
Conflict of Interest
ML I BH są pracownikami Topigs Norsvin Research Center, Instytutu Badawczego ściśle związanego z jednym z fundatorów (Topigs Norsvin).
wszyscy pozostali autorzy oświadczają, że badanie zostało przeprowadzone przy braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.
pozostali partnerzy Breed4Food Cobb Europe, CRV, Hendrix Genetics deklarują, że nie mają konkurencyjnych interesów w tym badaniu.,
podziękowania
autorzy dziękują Frankowi van Haarenowi i Toonowi Janssenowi za zorganizowanie komunikacji z gospodarstwem. Dziękujemy Manon Houben za jej wkład w badania patologiczne. Dziękujemy Moutowi Villerowi za zrobienie wideo i zdjęć chorym i zdrowym prosiaczkom po urodzeniu. Dziękujemy Jürgenowi Harliziusowi za opiekę nad prosiakami po przekazaniu. Dziękujemy Gerdzie van Eldik za pobranie próbek. Dziękujemy Egbertowi Knolowi i Egielowi Hanenbergowi za ogólne wsparcie i wskazówki ze strony Topigs-Norsvin.,
materiał uzupełniający
Video S1 | Video pokazujące obie chore osoby po urodzeniu.
Table S1 / Genomic variation in high LD with the ssc6 haplotype.
tabela S2 / genotypowanie miotu CxC z udziałem dwóch osobników dotkniętych chorobą (data porodu: 28 kwietnia 2019 r.). Tabela przedstawia homozygotyczny status del/del dla dwóch dotkniętych prosiąt 9912,9916.
tabela S3 / mapowanie i statystyki zasięgu z próbek WGS.
rysunek S1 | SPTBN4