Gen p53 został po raz pierwszy odkryty w 1979 roku. Białko zostało zidentyfikowane w komórkach myszy simian virus 40-przekształconych (SV40) przez immunoprecypitację z surowicą anty-T; białko to zostało nazwane białkiem p53 . W tym samym roku Kress i inni naukowcy odkryli nową klasę białek o masie cząsteczkowej w zakresie 50-60kda. Ten rodzaj białka został następnie zidentyfikowany jako p53 . Białko p53 można również zidentyfikować z różnych przekształconych linii komórkowych przez immunoprecypitację., Podobne wyniki Lane i Linzer osiągnęli również w 1979 roku. Innym dowodem na identyfikację p53 jest to, że p53 został wyrażony we wszystkich badanych przekształconych komórkach myszy; badania te obejmują indukowane chemicznie mięsaki, przekształcone fibroblasty i białaczki, podczas gdy w normalnych komórkach, p53 nie został wyrażony. Dodatkowo, wysoki poziom p53 został wykryty w większości przekształconych komórek, bez względu na to, jak komórki zostały przekształcone, spontanicznie lub nie spontanicznie ., Było to w dużej mierze spowodowane zwiększoną stabilnością p53, jednak w embrionalnych komórkach rakowiaka F9 wyrażał on wysoki poziom p53, wynikało to z ilości przetłumaczonego mRNA p53 .
po odkryciu białka p53 w 1979 roku, jego analiza stała się popularna. Jednak w tym czasie, ponieważ było to nowo odkryte białko i nie było dla niego dawnej nazwy, różne instytucje używały różnych nazw i publikowały artykuły o różnych nazwach., Aby rozwiązać ten problem, w 1983 roku, podczas i Międzynarodowych Warsztatów p53, które odbyły się w Oxted w Wielkiej Brytanii, naukowcy z różnych grup badawczych w różnych krajach spotkali się, aby omówić wspólną nomenklaturę dla tego nowo odkrytego białka. Na tym spotkaniu „p53” stała się jego nazwą i od tego czasu jest używana. Uważano, że powodem, dla którego naukowcy nazwali białko p53 jest to, że masa cząsteczkowa tego białka wynosi 53kDa, co opiera się na jego migracji w żelu SDS. Później okazało się, że masa cząsteczkowa jest błędna, a prawidłowa Masa cząsteczkowa powinna wynosić 43.,7kDa, ponieważ p53 zawiera region bogaty w prolinę, a region ten może zmniejszyć migrację p53 w żelu SDS. Ale nazwa „p53” pozostała .
w latach 80. uważano, że białko p53 bierze udział w cyklu komórkowym, a także odgrywa rolę w replikacji DNA. Później, w latach 1982-1994, ludzie odkryli, że niektóre wirusowe onkoproteiny były w stanie związać się z p53, tworząc kompleks. W 1982 roku Sarnow i in. stwierdzono, że adenowirus E1b (58kDa) może wchodzić w interakcje z białkiem 54kda obecnym w komórkach myszy przekształconych w SV40 wymienionych powyżej., Na podstawie wyników swoistości immunologicznej przeciwciał T oraz map peptydowych białka 54kDa, białko to jest identyfikowane jako p53 . W tym samym roku naukowcy odkryli, że jeśli wstrzyknęli przeciwciało p53 do Szwajcarskich komórek mysich 3T3, to zahamowałoby to wejście komórek w fazę s cyklu komórkowego; w tej samej sytuacji przeciwciało p53 nie miało wpływu na syntezę DNA wywołaną przez SV40 lub adenowirus .,
później w 1984 roku naukowcy zbadali wpływ p53 na nie przekształcone fibroblasty 3T3; przeanalizowali szybkość syntezy białka p53 w różnych punktach czasowych i odkryli, że w późnej fazie G1 szybkość syntezy i poziom białka p53 i związany z nim wzrost mRNA. Wynik ten sugeruje, że białko p53 hamuje wchodzenie komórek w fazę dzielącą z międzyfazą . Maltzman W et al. w tym samym roku przeprowadził kolejny eksperyment. Potraktowali nie przekształconą komórkę myszy światłem UV i promieniowaniem UV-mimetycznym chemicznym rakotwórczym 4nqo i wykryli wysoki poziom p53., Wynik pokazał, że podwyższona ekspresja p53 jest nie tylko symbolem, który wskazuje na cykl komórkowy, ale także, co ważniejsze, składnikiem, który bierze udział w syntezie DNA i proliferacji komórek . W 1987 roku, badając kompleks antygenu T wirusa simian 40 i polimerazy DNA α, Gannon i inni naukowcy odkryli podobną zmianę w antygenie po związaniu się z p53 i polimerazą α. Odkryli również, że przy pewnym stężeniu trzech składników mogą tworzyć specjalny kompleks trimeryczny, który zawiera antygen T, p53 i polimerazę DNA α., Ponieważ antygen T bierze udział w replikacji wirusowego DNA i transformacji komórkowej, wynik ten wskazuje, że p53 odgrywa rolę w kontroli cyklu komórkowego i replikacji DNA .
jak pokazano powyżej, p53 posiada zdolność do nieśmiertelności komórek. W 1984 roku Eliyahu D et al. okazało się, że p53 i produkt myc oncogene wspólne kilka podobnych właściwości. Oba mają zdolność wiązania się z innymi białkami i biorą udział w cyklu komórkowym, a oba gromadzą się w jądrach przekształconych komórek ., Bienz, Pennica i Oren przeanalizowali sekwencje aminokwasowe białka p53 i produktu myc i odkryli, że oba białka wykazują podobieństwa w strukturze molekularnej i pozycji specjalnych naładowanych reszt. Następnie naukowcy zaproponowali, że p53 może działać jako onkogen. Na podstawie tej hipotezy Eliyahu D et al. przeprowadziłem kilka eksperymentów. Ponieważ pierwszorzędowe fibroblasty zarodkowe mogą być przekształcane przez zaangażowanie zarówno produktu myc, jak i Ha-ras, pierwotne komórki nerek szczura dziecięcego mogą być również przekształcane przez współpracę Ha-ras i adenowirusa wczesnego regionu 1A, Eliyahu D et al., postanowił użyć tego rodzaju systemu badań biologicznych do identyfikacji onkogennej funkcji p53. Leczyli normalne komórki embrionalne p53 i aktywowali Ha-ras. Wynik wykazał, że komórki docelowe napotykają zmiany morfologiczne i produkują wysokie poziomy p53, Eliyahu D et al. uważa się, że transformacja embrionalnych fibroblastów przez p53 i Ha-ras wyjaśniła, że Gen p53 jest onkogenem ., W 1985 Jenkins zaproponował, że Gen p53 może wydłużyć żywotność komórek, zwiększyć afektywność transformacji poprzez zmianę sekwencji kodującej, która mogłaby spowodować produkcję stabilnych białek .
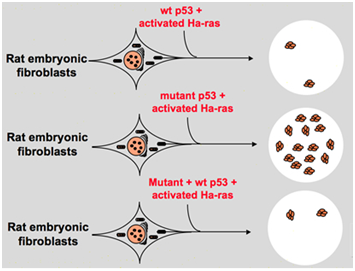
jednak pod koniec lat 80.naukowcy zaczęli zdawać sobie sprawę, że p53 jest genem supresorowym guza zamiast onkogenu. Zauważyli, że p53 z prawidłową funkcją nie może być wykryty w wielu guzach i okazało się, że utrata ekspresji i funkcji genu p53 typu dzikiego jest konieczna podczas transformacji komórkowej., Zwiększa to możliwość, że Gen typu dzikiego p53 może hamować progresję nowotworu . Następnie sformułowali inną hipotezę: Gen klonowy p53 używany w poprzednich eksperymentach zawiera dominujące negatywne mutacje w obrębie wysoce zachowanej domeny sporadycznie, co prowadzi do przeciwnych wyników eksperymentu . W 1988 roku Ben i inni naukowcy wykryli ogromną ilość przestawionych p53 w mysich liniach komórkowych erytroleukemii-DP20-1 i CB3, które pochodzą ze śledziony mysich zakażonych wirusem białaczki Friend ., W 1989 roku Eliyahu, który wskazał, że p53 jest onkogenem, zmienił zdanie i przypuszczał, że Gen typu dzikiego p53 może hamować transformację komórkową. Eliyahu i inni naukowcy badali wpływ białka p53 kodowanego przez plazmidy i zmutowanego p53 na zdolność wywoływania pierwotnej transformacji fibroblastów zarodkowych przez różne kombinacje onkogenów in vitro. Na przykład mutant p53 Plus ras i myc plus ras., Wynik wykazał, że p53 typu dzikiego prowadzi do ogromnej redukcji przekształconych ognisk wywołanych przez zmutowany p53 Plus ras; zmutowany p53 nie hamuje przekształconych ognisk wywołanych przez myc Plus ras, podczas gdy transformacja za pośrednictwem myc Plus ras jest bardzo wrażliwa na ekspresję p53 typu dzikiego. Rysunek 1 pokazuje ten eksperyment zwięźle. Wykazano, że w porównaniu z mutantem p53, dziki Typ p53 wykazuje wyraźnie hamujący wpływ na transformację komórkową. Efekt jest dodatnio związany z poziomem ekspresji mutanta p53 typu dzikiego i ujemnie związany z poziomem ekspresji mutanta p53., Ten eksperyment sugerował, że dziki Typ p53 może rzeczywiście mieć odwrotną funkcję w porównaniu z zmutowanym p53 i może hamować nowotwór . Obecnie p53 jest rozpoznawany jako gen supresorowy guza. Szacuje się, że około połowa guzów jest wywoływana przez p53. Jest jednym z najczęściej mutowanych genów u ludzi i najczęściej analizowanym genem na całym świecie .
w ciągu pierwszych kilku lat lat 80. biochemiczny szlak p53 i efekt mutacji p53 nie były jasne. W 1991 roku Kern i inni naukowcy odkryli, że sekwencja DNA 33-zasadowa wiąże się specyficznie z dzikim typem p53 in vitro. Stwierdzono również, że białko p53 zawiera dwie mutacje, które zwykle występują w guzach ludzkich, które nie mogą wiązać się z tym specyficznym regionem DNA. Przypuszczali więc, że funkcja p53 zależy od jego zdolności do wiązania określonych sekwencji DNA, a zdolność ta jest zmieniana przez mutacje Znalezione w ludzkich guzach., Przypuszczają również, że ta sekwencja DNA pary 33 zasad może nie być jedyną sekwencją, która ma zdolność wiązania się specyficznie z p53 u ludzi; jednak może pomóc ludziom lepiej zrozumieć funkcję p53 . Później stwierdzono, że p53 odgrywa rolę podczas cyklu komórkowego, naprawy DNA, różnicowania, inicjowania apoptozy i angiogenezy. Rotter V et al. stwierdzono, że p53 up-reguluje różnicowanie komórek. Na przykład wysoki poziom białka p53 został wykryty w kilku kluczowych etapach podczas różnicowania komórek B. Podwyższone p53 można również wykryć podczas spermatogenezy., Tymczasem tylko bardzo niski poziom białka p53 można wykryć w niektórych narządach dorosłych myszy .
w 1990 roku okazjonalnie odkryto przydatne narzędzie. Jest to wrażliwy na temperaturę mutant p53, zwany p53val135. Może działać jako prawdziwy Dziki Typ p53 w temperaturze 32,5 oC, hamując transformację, a także może działać jako inny zmutowany p53 w temperaturze 37,5 oC lub powyżej 48oC, wywołując transformację. Dodatkowo, dla przekształconych komórek wyrażających p53val135, jego proliferacja jest kontrolowana w temperaturze permisywnej, a ten rodzaj kontroli jest odwracalny., Za pomocą tego mutanta p53val135 odkryto, że dziki Typ p53 wywołuje zatrzymanie cyklu komórkowego przy G1 lub G2 / M . W 1991 Elisheva et al. stwierdzono, że wrażliwy na temperaturę p53val135 pełni inną funkcję w linii komórek mysich białaczek szpikowych. Po reaktywacji p53val135 przez kilka dni wszystkie komórki obumarły, a śmierć ta wykazuje pewne właściwości apoptozy . Rok później podobny wynik uzyskał Shaw. Dziki Typ p53 został przetransferowany do ludzkiej linii komórkowej nowotworu jelita grubego EB., Komórki zostały zbadane pod mikroskopem świetlnym i elektronowym i stwierdzono, że wykazują pewne właściwości apoptozy . W 1990 roku Scheffner et al. inni naukowcy odkryli, że E6, który stymuluje niszczenie białek regulacyjnych komórek gospodarza, jest kodowany przez onkogennego wirusa brodawczaka ludzkiego typu 16 i 18 i może tworzyć kompleks z dzikim typem p53 in vitro, co z kolei powoduje degradację białka p53 .
w 1992 roku odkryto kluczowe białko MDM2, ponieważ wiąże się ono ściśle z p53 i hamuje transaktywację za pośrednictwem p53., Masa cząsteczkowa MDM2 wynosi 90kDa i tworzy kompleks z zarówno zmutowanym, jak i dzikim typem p53 . W tym samym roku Livingstone RL et al. zbadano, czy komórka utraciła jedną lub obie kopie alleli dzikiego typu p53 i czy było to wystarczające do amplifikacji genów. Amplifikację genów Wykryto głównie w przekształconych komórkach, ale nie w normalnych fibroblastach. Wynik wykazał, że komórki tracące jedną kopię alleli p53 działają jak dzikie p53, podczas gdy komórki tracące obie kopie alleli p53 wykazują większą częstotliwość amplifikacji . Kolejny eksperyment wykonany przez Yin Y et al., podobne wyniki .
w 1993 roku zidentyfikowano Gen docelowy p53 o nazwie CDKN1A. Koduje białko p21, które jest inhibitorem kinazy zależnej od cyklin, które hamuje cyklinę-CDK2 i CDK1 przez wiązanie się z nimi. W 1993 Szekely odkrył, że antygen jądrowy wirusa Epsteina-Barr 5 (EBNA-5) jest kodowany przez wirus Epsteina-Barr i może zainfekować ludzką komórkę limfoblastoidalną B. Peptyd o długości 66 aminokwasów jest odpowiedzialny za tworzenie kompleksu EBNA-5-p53, mutacje punktowe p53 nie wpływają na jego zdolność wiązania z EBNA-5., Hamuje jednak powstawanie kompleksów z innymi cząsteczkami . W 1994 cho i jego współpracownicy po raz pierwszy opisali strukturę krystaliczną kompleksu P53-DNA. Ta domena wiążąca DNA była również nazywana domeną rdzeniową. Zawiera pozostałości 102-292 i składa się z kanapki beta. Zademonstrowali również szczegółową strukturę podstawowej domeny . Również w 1994 r. Wang XW et al. interakcje pomiędzy białkiem x wirusa zapalenia wątroby typu B (HBX) a białkiem p53 typu dzikiego u ludzi., Odkryli, że HBX może hamować zdolność p53 do wiązania się z innym DNA specyficznym dla sekwencji po jego związaniu z p53 i może również hamować skojarzenie p53 z czynnikami transkrypcyjnymi .
w 1997 roku Honda R i wsp. po raz pierwszy postawiono hipotezę, że MDM2 może wywołać ubikwitylację p53 i prowadzić do degradacji p53 przez układ ubikwitynowo-proteasomowy. Wskazali, że MDM2 wiąże się z domeną N-terminalną (NTD) p53 i działa jako ligaza ubikwitynowa E3 . Również w 1997 roku odkryto dwie nowe rodziny białek, p63 i p73, które mają znaczącą homologię z p53., p73, zwane również białkiem nowotworowym 73, jest kodowane przez gen znajdujący się w 1p36. Lokalizacja jest usuwana często w neuroblastoma i innych guzów. p73 może aktywować geny docelowe p53 i oddziaływać z p53 . Yang et al. stwierdzono, że Gen p63 znajduje się w 3q27-29 i może być wykryty w różnych komórkach myszy i ludzi. Podobnie jak p73, p63 może znacznie transactivate genów docelowych p53, może również indukować apoptozę. Jedną z cech p63 jest to, że większość p63 nie ma N-końca ., W tym samym roku Serrano i współpracownicy odkryli, że pierwotne mysie fibroblasty mogą być przekształcane przez ras w przypadku braku p53 lub p16, a nieaktywne p53 lub p16 mogą ułatwić proces nieśmiertelności ludzkich komórek. Wyniki te sugerują, że p53 odgrywa rolę w starzeniu się komórek . Następnie, w 1997, stwierdzono, że p53 odgrywa rolę w inicjacji apoptozy. Gdy komórki wchodzą w fazę proliferacji, telomery na końcu każdego chromosomu skracają się po każdej rundzie replikacji DNA z powodu niekompletnej replikacji pojedynczego pasma DNA na końcu podstawki DNA ., Aktywowany Gen supresorowy guza p53 ogranicza liczbę przypadków podziału komórek. Wynford TD odkrył, że wraz z utratą funkcji dzikiego typu p53 wszystkie fibroblasty uciekają przed apoptozą. Ponadto funkcja transactivation p53 może być włączona przez apoptozę . Wynford TD zaproponował, że istnieją trzy możliwości aktywacji p53. Pierwszy to modyfikacja potranslacyjna przez fosforylację, drugi to up-reguluje kofaktory nadkrytyczne, takie jak p33ING1, ostatni to down-reguluje inhibitory p53, takie jak MDM2 .
, badał cele transkrypcyjne p53 u Drosophila. Istnieją dowody na to, że oczy Drosophila wykazują ciężki fenotyp szorstkiego oka pod ekspresją ludzkiego p53, który indukuje apoptozę wyobrażeniowych komórek dysku oka, powodując utratę komórek pigmentowych, wreszcie hamując rozwój Drosophila, więc Drosophila może być zwierzęciem modelowym do badania funkcji p53. Brodsky odkrył, że Gen rpr zawiera konsensus p53 miejsce wiązania, które znajduje się w regionie cis-regulacji rpr, a także jest aktywatorem apoptozy., Z innymi dowodami Brodsky twierdził, że rpr jest jednym z celów transkrypcyjnych p53 . W 2001 roku Derry i współpracownicy odkryli, że C. elegans nie ma genu p53, ale rzeczywiście zawiera Gen CEP-1 kodujący białka o podobnej sekwencji z białkiem p53. Ten gen C. elegans koduje białko CEP-1, które ma zdolność wywoływania apoptozy przez stres genotoksyczny i jest niezbędnym składnikiem podczas mejozy .
w 2002 Tyner i współpracownicy zaproponowali, że p53 odgrywa rolę w regulacji starzenia się organizmów., W celu zbadania funkcji p53, stworzyli genetycznie zmodyfikowane myszy ze zmutowanym p53 przez usunięcie eksonów 1-6 i regionu upstream dzikiego typu genu p53 (p53+/+), zwanego p53+/m. działa on jako dzikiego typu p53 i ma zwiększoną odporność na spontaniczne guzy lepiej niż dzikiego typu p53. W eksperymencie monitorowano myszy zawierające p53+ / m, p53+ / + i p53+/ -. p53 + / – oznacza, że myszy tracą jedną kopię genu p53 typu dzikiego., Wyniki wykazały, że żadna z myszy z p53 + / m nie rozwinęła guzów zagrażających życiu, jednak ponad 80% myszy z p53 + / – i ponad 45% myszy z p53+ / + rozwinęło tego rodzaju guzy. Patrząc wewnątrz guzów, miejscowe zmiany nowotworowe obserwowano u 2 z 35 myszy p53 + / m, natomiast różne guzy, takie jak chłoniaki i kostniakomięsaki stwierdzono u myszy p53+/- i p53+/+. Podczas tego eksperymentu zaobserwowano również, że mediana wieku p53+ / m wynosiła 96 tygodni, podczas gdy mediana wieku p53 + /m wynosiła 116 do 118 tygodni., Tyner i współpracownicy zbadali również możliwość, że krótsza żywotność p53+ / m jest związana ze starzeniem się. Okazało się, że po 18 miesiącach myszy p53+/m zaczęły tracić na wadze i wigorze, tak jak w przypadku myszy p53+/m obserwowano zmniejszoną masę ciała w wieku 30-36 miesięcy. myszy p53+ / m wykazują również lordokyphosis. W zależności od analizy rentgenowskiej myszy p53+ / m wykazywały zmniejszoną gęstość kości w wieku 12 miesięcy i staną się ciężkie w wieku 18 miesięcy. Jest to symbol osteoporozy i osteoporoza jest markerem starzenia się u ludzi i myszy . Tyner i in., przetestowano również tolerancję stresu, ponieważ zdolność ta jest również markerem starzenia się . Wykonali biopsje uderzeniowe 3 mm w skórę pleców starych i młodych znieczulonych myszy p53+/m i p53+/+. Ich wyniki wykazały, że wiele starych myszy p53+/m zmarło po wstrzyknięciu standardowej dawki Avertyny, co wskazuje, że stare myszy p53+/m były mniej tolerancyjne na stres .
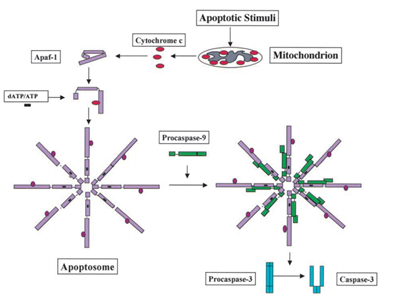
w 1991 r .stwierdzono, że p53 ma zdolność wywoływania apoptozy, natomiast w 2003 r. Mihara i inni naukowcy odkryli, że p53 pełni również rolę apoptozy w mitochondriach., Ponieważ niektóre białka mitochondrialne mają zdolność aktywowania apoptozy komórkowej poprzez aktywne kaspazy lub neutralizowanie inhibitorów cytozolowych. W przykładzie kaspazy indukowanej przez cytochrom c, po otrzymaniu sygnału apoptozy, cytochrom c jest uwalniany z przestrzeni międzycząsteczkowej mitochondriów, a następnie wiąże się z Apf-1, który istnieje jako nieaktywny monomer, indukuje jego zmianę konformacyjną i zwiększa powinowactwo wiązania do dATP/ATP 10-krotnie niż Apaf-1 wiąże sam dATP/ATP. Następnie kompleks Apaf-1-cytochromu c wiąże się z DATP / ATP, tworząc apoptosom., Następnie domena rekrutacyjna caspazy (CARD) Apaf-1 ujawniła się w apoptosomie, rekrutuj procaspazę-9, a następnie autoaktywuj się. Końcowy kompleks rozszczepia się i aktywuje inne kaspazy, takie jak kaspaza-3, które z kolei rozszczepią ważne cząsteczki w komórce, powodując kondensację chromatyny, fragmentację DNA i ostatecznie prowadząc do apoptozy . Rysunek 2 przedstawia szlak aktywacji kaspazy indukowanej przez cytochrom C.
naukowcy odkryli, że Gen typu dzikiego p53 może być szybko translokowany do powierzchni mitochondrialnej komórek nowotworowych. W eksperymencie okazało się, że niektóre wywołane stresem białko p53 typu dzikiego ma zdolność do translokacji do mitochondriów tymocytów w komórkach ludzkich lub mysich po apoptozie z powodu uszkodzenia DNA i niedotlenienia. Następnie te białka typu dzikiego p53 indukują permeabilizację mitochondriów i powodują szereg zmian zachodzących w mitochondriach, takich jak uwalnianie cytochromu c, tworząc kompleks z Bcl2 i BclXL .,
jako dobry wynik kliniczny z niewielkim efektem ubocznym, terapia genowa jest popularna. Do końca 2005 roku w bazie Journal of Gene Medicine było 1020 badań terapii genowej. Wśród tych badań, 66% terapii genowych przeprowadzono na chorych na raka, a 58 badań tego używanego rAd-p53, rekombinowanego adenowirusa kodującego ludzki gen p53. W kwietniu 2004 oficjalnie uruchomiono rekombinowany ludzki adenowirus-p53 injection (Gendycyna). Gendycyna jest stosowana w leczeniu raka płaskonabłonkowego głowy i szyi i została zatwierdzona przez Państwową Agencję Żywności i Leków Chin w październiku. 16, 2003., Stał się pierwszym produktem terapii genowej na świecie, które zostały zatwierdzone przez chiński rząd .
w 2005 roku odkryto, że Gen p53 reguluje metabolizm. Aby przenieść się z fazy G1 do fazy S, komórki muszą mieć wystarczające wsparcie surowców dla DNA, organelli i syntezy białek. Aby uregulować ten proces, konieczne są pewne punkty kontrolne. Jednym z nich jest zależny od glukozy punkt kontrolny w G1 / S. jest regulowany przez kinazę białkową aktywowaną AMP (AMPK). Po wyczerpaniu glukozy AMPK może fosforylować białko p53, co z kolei indukuje zatrzymanie komórek i zapobiega ich śmierci., Komórki, które napotkają zatrzymanie zależne od p53, ponownie wprowadzą cykl komórkowy, gdy glukoza będzie wystarczająca .
wiadomo, że inaktywacja p53 jest konieczna do powstawania nowotworów. Bykow i in. VJ i Snydel el et al. zwróć uwagę, że niewłaściwe funkcjonowanie p53 może prowadzić do proliferacji istniejącego guza . Ventura i jego współpracownicy przeprowadzili kilka eksperymentów, aby przetestować tę hipotezę. Przywrócili funkcję endogennego p53 w pierwotnych guzach autochtonicznych w celu zbadania konsekwencji reaktywacji p53., Wynik wykazał, że reaktywacja p53 była odpowiedzialna za regresję guzów autochtonicznych. Oznacza to, że inaktywowane białko p53 może prowadzić do rozwoju guza. Xue i inni naukowcy przeprowadzili również eksperyment, aby przetestować konsekwencje reaktywacji p53 na guzach. Wykorzystali odwracalną interferencję RNA (RNAi) do regulacji ekspresji endogennego p53 u myszy z rakiem wątroby. W eksperymencie doksycyklina (Dox) jest używana do reaktywacji p53, ponieważ ekspresja p53 jest całkowicie tłumiona, gdy brakuje Dox i szybko przywracana po dodaniu Dox., Podczas leczenia Dox, miRNA p53 została wyłączona, co z kolei powoduje zwiększoną ekspresję p53. Wynik wykazał, że guzy u myszy leczonych Dox stają się niewykrywalne po 12 dniach, podczas gdy guzy u nieleczonych myszy rosły szybko. Aby sprawdzić następstwo przejściowej reaktywacji p53, leczono myszy Dox przez 4 dni, a następnie przerwano. Wynik wykazał, że nawet dwudniowe leczenie może powodować regresję guzów, a 4 dni leczenia może spowodować całkowitą regresję guzów., Wskazali również, że podczas regresji nowotworu reaktywowany przejściowo p53 może wywołać starzenie się komórek, a nie apoptozę. W tym samym roku Hu odkrył, że implantacja zarodkowa u samic myszy p53-/- jest regulowana przez czynnik hamujący białaczkę (Lif). LIF jest wydzielaną cytokiną i jest ważny dla implantacji blastocysty. Gen kodujący LIF jest identyfikowany jako gen docelowy p53, a miejsce wiązania p53 znajduje się w intronie 1 u ludzi i myszy .