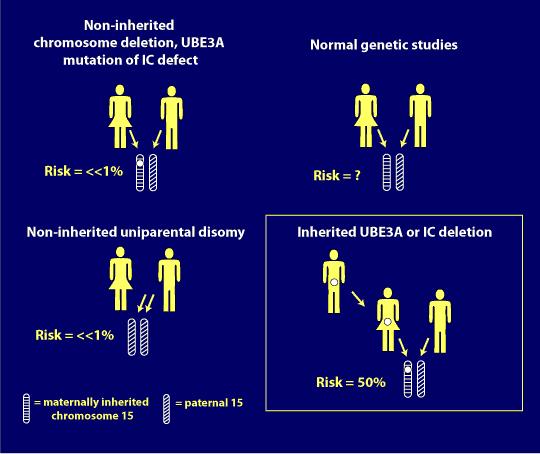
alvorlighed af genotyper
desværre har den mest almindelige som genotype, deletion positive, en tendens til at være den mest alvorlige, hvad angår symptomer eller egenskaber.
Nedenfor er en illustration Dr. Charles Williams brugte i sin genetiske præsentation i 2014 Canadiske Angelman Syndrom Organisation (CASS) Konference for at vise forholdet mellem SOM genotyper og sværhedsgraden af nogle karakteristiske træk.,
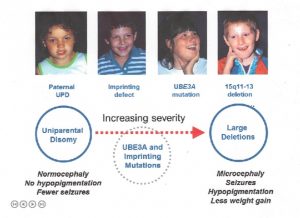
Risiko for Recidiv i Angelman Syndrom
En genetisk rådgiver kan informere dig om muligheden for Angelman syndrom opstår eller gentager sig gennem indsamling af familiens historie og blodprøver. Følgende oplysninger kan være nyttige til at forstå den genetiske risiko for Angelman syndrom, men er ikke beregnet til at erstatte genetisk rådgivning.
1., Fælles kromosomdeletion:
Mere, at 98% af kromosomdeletion tilfælde opstå ved en spontan begivenhed, og de er således ikke arvet; recidiv risikoen er <<1% for disse familier. Imidlertid forekommer 1-2% af sletninger på grund af en arvelig abnormitet i moderkromosomet 15, såsom en afbalanceret kromosomtranslokation. En anden meget lille gruppe (f. eks.,, kun få tilfælde rapporteret i litteraturen), kan skyldes en meget lille, maternelt arvet kromosomsletning, der involverer et lille område omkring og inklusive UBE3A-genet. I disse tilfælde øges risikoen for tilbagefald af moderen afhængigt af den tilstedeværende abnormitet. Kromosomundersøgelse af moderen, inklusive fisk, hjælper med at udelukke arvelige kromosom 15-abnormiteter.
2. Faderlig uniparental disomi (patUPD):
mere end 99% af patUPD-tilfælde forekommer som en tilsyneladende spontan, ikke-arvet begivenhed., Hvis en person har SOM følge af patUPD og har en normal karyotype, en kromosom-analyse af moderen bør ikke desto mindre blive tilbudt med henblik på at udelukke den sjældne mulighed for, at en Robertsonsk translokation eller markør kromosom var en disponerende faktor (fx via generation af mødres gamet, at der var nullisomic for kromosom 15, med efterfølgende post-zygotic “korrektion” at fædrene disomy).
3. Imprinting Center (IC) defekt:
der er to typer af IC defekter: sletninger og ikke-sletninger., Ikke-sletningshændelser ser ikke ud til at være arvet og har en <1% gentagelsesrisiko. De fleste sletninger er ikke arvet, men en betydelig del af dem er (dvs.moderligt arvet), og disse giver en 50% risiko for gentagelse.
4. UBE3A-mutationer:
UBE3A-mutation kan enten forekomme spontant (f.eks. ikke arvet og uden øget tilbagefaldsrisiko) eller være maternalt arvet og have en 50% risiko for gentagelse (se nedenfor for prægning af arv).
5., Enkeltpersoner, der ikke har kendt mekanisme (alle 4 ovenstående mekanismer er blevet fjernet):
For forældre SOM personer, der har tilsyneladende normal genetiske tests (ingen beviser for sletning, prægning defekt, UPD eller UBE3A mutation), og dermed deres børn er kun klinisk diagnosticeret, det vides ikke, hvad recidiv risikoen er. En øget risiko synes sandsynlig, men overstiger sandsynligvis ikke 10%.
6. Kimcellemosaik:
dette udtryk henviser til et fænomen, hvor en genetisk defekt er til stede i cellerne i gonaden (æggestok i moderens tilfælde), men ikke i andre celler i kroppen., Denne forekomst kan føre til fejl i risikovurderingen, fordi en genetisk test, for eksempel på en mors blodlegemer, vil være normal, når der faktisk er en genetisk defekt i kimcellerne i hendes æggestokk. Heldigvis forekommer kimcellemosaik meget sjældent. Ikke desto mindre er det blevet observeret i som forårsaget af mekanismerne for stor kromosom deletion, Imprinting Center deletion og UBE3A mutation.
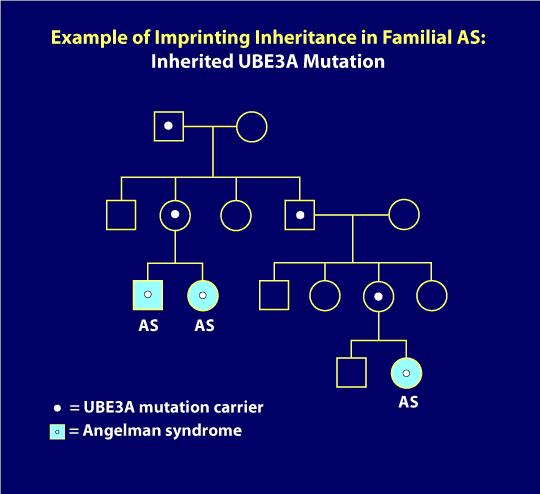
7., Prægning arv:
UBE3A mutationer og Prægning Center sletninger kan udstille prægning arv, hvor et luftfartsselskab far kan passere på den genetiske defekt er til hans børn, uden at det skaber problemer, men når en kvinde passerer den samme genetiske defekt videre til sine børn, uanset kønnet på sit barn, at barnet vil have med. Stamtavle diagrammet nedenfor illustrerer prægning arv. Her, som det først er sket, efter at en bærermor overførte genfejlen (for eksempel som i de to søskende med som afbildet på venstre nederste del af stamtavlen)., Derudover har en fjern fætter i denne familie også som følge af den prægende arv. I diagrammet har personer med de lyseblå cirkler eller firkanter som men alle andre i familien er klinisk normale. De hvide prikker repræsenterer asymptomatiske, normale bærere af AS-mutationen. Når en as-genetisk mekanisme bestemmes at blive arvet, kan genetisk test af familiemedlemmer normalt identificere bærere af genfejlen. Som du måske forestiller dig, rådes professionel genetisk rådgivning i disse situationer.