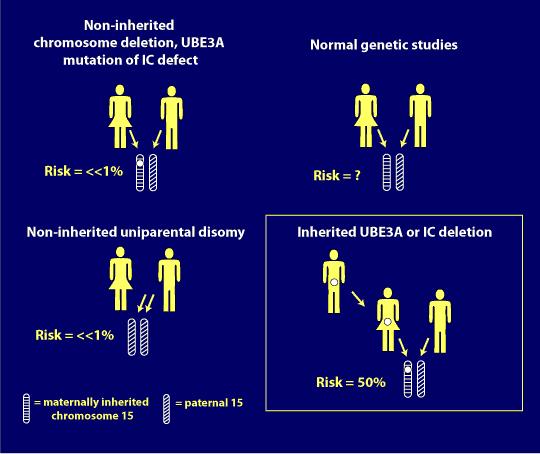
nasilenie genotypów
Niestety, najczęstsze genotyp AS, delecja dodatnia, wydaje się być najcięższa, jeśli chodzi o objawy lub cechy.
Poniżej znajduje się ilustracja, której dr Charles Williams użył w swojej prezentacji genetycznej na konferencji Canadian Angelman Syndrome Organization (CASS) w 2014 roku, aby pokazać związek między genotypami AS a ciężkością niektórych jako cech.,
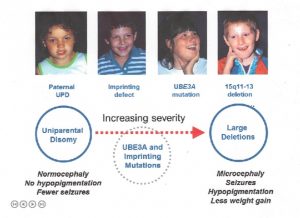
ryzyko nawrotu w zespole Angelmana
doradca genetyczny może poinformować Cię o możliwości wystąpienia zespołu Angelmana lub nawrotu poprzez zbieranie historii rodziny i badania krwi. Poniższe informacje mogą być pomocne w zrozumieniu ryzyka genetycznego zespołu Angelmana, ale nie ma na celu zastąpienia poradnictwa genetycznego.
1., Częsta delecja chromosomów:
Więcej, że 98% przypadków delecji chromosomów występuje w wyniku spontanicznego zdarzenia, a zatem nie jest dziedziczone; ryzyko nawrotu wynosi< < 1% dla tych rodzin. Jednak 1-2% delecji występuje z powodu dziedzicznej nieprawidłowości w chromosomie 15, takich jak zrównoważona translokacja chromosomu. Inna bardzo mała grupa (np.,, tylko kilka przypadków opisywanych w literaturze), może być spowodowane bardzo małą, dziedziczną delecją chromosomów, która obejmuje niewielki obszar wokół i obejmujący Gen UBE3A. W tych przypadkach ryzyko nawrotów u matki jest zwiększone w zależności od rodzaju występującej nieprawidłowości. Badanie chromosomu matki, w tym ryb, pomaga wykluczyć dziedziczne nieprawidłowości chromosomu 15.
2. Patupd (paternal uniparental disomy):
ponad 99% przypadków patUPD występuje jako pozorne spontaniczne, nie dziedziczone Zdarzenie., Jeśli dana osoba ma as z powodu patUPD i ma prawidłowy kariotyp, analiza chromosomalna matki powinna być jednak oferowana w celu wykluczenia rzadkiej możliwości, że translokacja Robertsona lub chromosom markera była czynnikiem predysponującym (np. poprzez generację gamety matki, która była zerowa dla chromosomu 15, z późniejszą „korekcją” pożygotyczną do dysomii ojcowskiej).
3. Defekt centrum imprintingu (IC):
istnieją dwa rodzaje defektów IC: delecje i brak delecji., Zdarzenia bez delecji nie wydają się być dziedziczone i mają <1% ryzyko nawrotu. Większość delecji nie jest dziedziczona, ale znaczna ich część (tj. dziedziczona Matczynie), a te dają 50% ryzyko nawrotu.
4. Mutacje UBE3A:
mutacja UBE3A może wystąpić spontanicznie (np. nie dziedziczona i bez zwiększonego ryzyka nawrotu) lub być dziedziczona Matczynie i mieć 50% ryzyko nawrotu(patrz poniżej w przypadku dziedziczenia z nadrukiem).
5., Osoby z nieznanym mechanizmem (wszystkie 4 powyższe mechanizmy zostały wyeliminowane):
dla rodziców osób, które mają widoczne normalne testy genetyczne (brak dowodów na delecję, defekt nadruku, mutację UPD lub UBE3A), a więc ich dzieci są zdiagnozowane tylko klinicznie, nie wiadomo, jakie jest ryzyko nawrotu. Zwiększone ryzyko wydaje się prawdopodobne, ale prawdopodobnie nie przekracza 10%.
6. Mozaika komórek zarodkowych:
termin ten odnosi się do zjawiska, w którym wada genetyczna występuje w komórkach gonady (jajniku w przypadku matki), ale nie w innych komórkach organizmu., Wystąpienie to może prowadzić do błędów w ocenie ryzyka, ponieważ badanie genetyczne, na przykład na komórkach krwi matki, będzie normalne, gdy w rzeczywistości wada genetyczna jest obecna w komórkach germinalnych jej jajnika. Na szczęście mozaika komórek zarodkowych występuje bardzo rzadko. Niemniej jednak zaobserwowano, że jest to spowodowane mechanizmami delecji dużych chromosomów, delecji Centrum imprintingu i mutacji UBE3A.
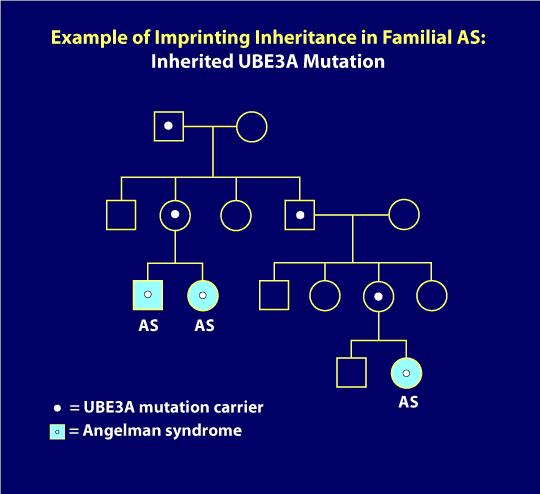
7., Dziedziczenie Imprinting:
mutacje UBE3A i delecje Imprinting Center mogą wykazywać dziedziczenie imprinting, w którym ojciec nosiciela może przekazać wadę genetyczną swoim dzieciom bez powodowania żadnych problemów, ale za każdym razem, gdy kobieta przekazuje tę samą wadę genetyczną swoim dzieciom, niezależnie od płci jej dziecka, to dziecko będzie miało AS. Poniższy diagram rodowodu ilustruje nadruk dziedziczenia. Tutaj, jak miało to miejsce dopiero po przekazaniu przez matkę nosicielkę wady genetycznej (np. jak u dwójki rodzeństwa z jak na zdjęciu po lewej dolnej części rodowodu)., Ponadto, daleki kuzyn w tej rodzinie ma również jako ze względu na dziedziczenie imprinting. Na diagramie osoby z jasnoniebieskimi kółkami lub kwadratami mają Jak, ale wszyscy inni w rodzinie są klinicznie normalni. Białe kropki reprezentują bezobjawowe, normalne nosiciele mutacji AS. Gdy genetyczny mechanizm AS jest określony jako dziedziczny, badania genetyczne członków rodziny mogą zazwyczaj zidentyfikować nosicieli wady genu. Jak można sobie wyobrazić, profesjonalne doradztwo genetyczne jest zalecane w takich sytuacjach.