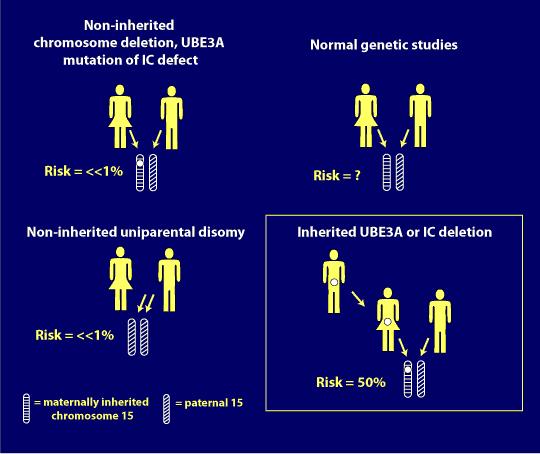
a genotípusok súlyossága
sajnos a genotípus, a deléció pozitív, a tünetek vagy jellemzők szempontjából a legsúlyosabb.
Az alábbiakban egy illusztráció, amelyet Dr. Charles Williams a 2014-es kanadai Angelman-szindróma szervezet (CASS) konferencián genetikai előadásában használt, hogy megmutassa az AS genotípusok közötti kapcsolatot és egyesek súlyosságát, mint jellemzőket.,
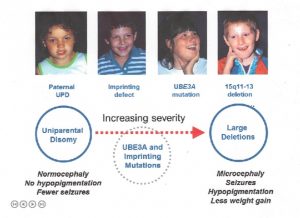
az Angelman-szindróma megismétlődésének kockázata
egy genetikai tanácsadó tájékoztathatja Önt arról, hogy az Angelman-szindróma előfordulhat vagy megismétlődhet a családi anamnézis és a vérvizsgálat összegyűjtése révén. Az alábbi információk hasznosak lehetnek az Angelman-szindróma genetikai kockázatának megértésében, de nem célja a genetikai tanácsadás helyettesítése.
1., Gyakori kromoszóma deléció:
Több, hogy a kromoszóma deléciós esetek 98% – a spontán esemény következtében alakul ki, és így nem öröklődik; a kiújulási kockázat <<1% ezeknél a családoknál. A törlések 1-2% – A azonban az anyai 15 kromoszóma öröklött rendellenessége, például a kiegyensúlyozott kromoszóma transzlokáció miatt következik be. Egy másik nagyon kis csoport (pl.,, csak néhány esetben számoltak be a szakirodalomban), lehet, mivel a nagyon kicsi, maternálisan öröklött kromoszóma deléció, amely magában foglalja egy kis terület körül, beleértve az UBE3A gén. Ezekben az esetekben az anyai kiújulás kockázata a jelen lévő rendellenesség típusától függően növekszik. Az anya kromoszóma vizsgálata, beleértve a halakat is, segít kizárni az örökölt 15.kromoszóma rendellenességeket.
2. Apai uniparental disomy (patUPD):
a patUPD esetek több mint 99%-a nyilvánvaló spontán, nem örökölt eseményként fordul elő., Ha egy személy miatt, MINT patUPD, s egy normális, kariotípus, egy kromoszóma analízis, az anya azonban felajánlotta, annak érdekében, hogy kizárja a ritka lehetőséget, hogy egy Robertsonian transzlokáció vagy a jelölő kromoszóma volt hajlamosító tényező (pl. keresztül generációs anyai ivarsejt volt nullisomic a kromoszóma 15, a későbbi post-zygotic “korrekció”, hogy apai disomy).
3. Imprinting Center (IC) hiba:
kétféle IC hibák: törlések és nem törlések., Úgy tűnik, hogy a nem-deléciós események nem öröklődnek, és <1% – os kiújulási kockázattal járnak. A legtöbb deléció nem öröklődik, de jelentős részük (azaz anyai öröklődés), és ezek 50% – os kockázatot jelentenek a kiújulásra.
4. UBE3A mutációk:
az UBE3A mutáció spontán módon (pl. nem öröklődik, és nem áll fenn a kiújulás kockázata), vagy anyára öröklődik, és 50% – os a kiújulás kockázata(lásd alább a lenyomat öröklés).
5., Ismert mechanizmussal nem rendelkező egyének (mind a fenti 4 mechanizmus megszűnt):
olyan egyének szülei esetében, akiknek nyilvánvaló normál genetikai tesztje van (nincs bizonyíték törlésre, imprinting hibára, UPD vagy UBE3A mutációra), és így gyermekeiket csak klinikailag diagnosztizálják, nem ismert, hogy mi a megismétlődési kockázat. A megnövekedett kockázat valószínűnek tűnik, de valószínűleg nem haladja meg a 10% – ot.
6. Csírasejt mozaicizmus:
Ez a kifejezés olyan jelenségre utal, amelyben genetikai hiba van jelen a gonád sejtjeiben (petefészek az anya esetében), de nem a test más sejtjeiben., Ez az esemény a kockázatértékelés hibáihoz vezethet, mivel a genetikai teszt, például egy anya vérsejtjein, normális lesz, ha valójában genetikai hiba van jelen a petefészek csíravonal-sejtjeiben. Szerencsére a csírasejtes mozaicizmus nagyon ritkán fordul elő. Mindazonáltal, azt figyelték meg, mint okozta mechanizmusok nagy kromoszóma deléció, Imprinting Center deléció és UBE3A mutáció.
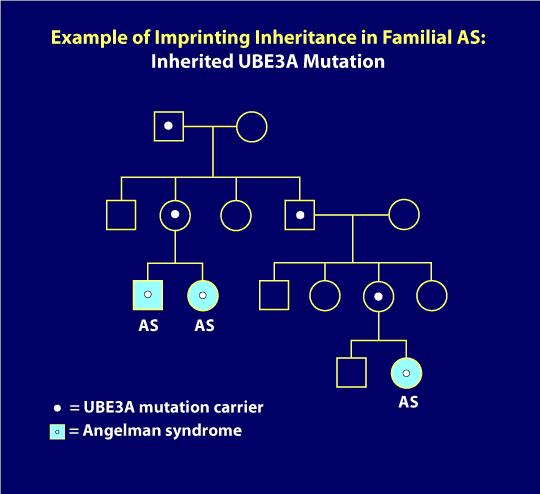
7., Feltöltési örökség:
UBE3A mutációk, illetve Feltöltési Center törléseket kiállítás feltöltési örökség, ahol a fuvarozó apja átadja a genetikai hiba, hogy a gyerekeknek nem okoz semmilyen problémát, de ha egy nő áthalad ugyanezt a genetikai hiba, hogy a gyerekek, függetlenül attól, hogy a szex a gyerekét, az a gyerek lesz, MINT. Az alábbi törzskönyvi ábra szemlélteti az öröklést. Itt, mint csak akkor történt, miután egy hordozó anya átadta a génhibát (például a két testvérnél, mint a törzskönyv bal alsó részén látható képen)., Ezen túlmenően, egy távoli unokatestvére ebben a családban is, mivel a imprinting örökséget. Az ábrán, egyének a világoskék körök vagy négyzetek, mint de mindenki más a családban klinikailag normális. A fehér pontok az AS mutáció tünetmentes, normál hordozóit képviselik. Ha az AS genetikai mechanizmust öröklésre határozzák meg, a családtagok genetikai vizsgálata általában azonosítja a génhiba hordozóit. Mint gondolnád, ezekben a helyzetekben tanácsos szakmai genetikai tanácsadás.