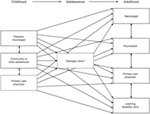
Figure 2. Schéma illustrant la dispersion croissante de l’offre de soins après le passage des services pédiatriques aux services pour adultes (3). Reproduit avec la permission de John Libbey Eurotext.,
autres considérations pour la prise en charge du SGL
Les patients atteints du syndrome de Lennox–Gastaut sont altérés dans leur vie quotidienne non seulement par divers types de crises qui sont souvent fréquentes et physiquement dommageables (comme les crises de goutte), mais aussi par diverses comorbidités graves (3). Les comorbidités qui sont particulièrement associées à LGS comprennent des problèmes cognitifs et comportementaux, un handicap physique et des troubles du sommeil., Cinq ans après le début de LGS, 75-95% des patients ont une déficience cognitive (101, 102), et des problèmes de comportement, tels que l’hyperactivité, l’agressivité et les traits autistiques, se développent dans environ 50% (10). La mobilité est souvent gravement affectée par les crises fréquentes (en particulier les crises de chute), qui sont physiquement exigeantes et entraînent souvent des blessures (3). Les Patients doivent souvent utiliser un équipement de protection (comme un fauteuil roulant, un casque et/ou un masque facial) pour minimiser les effets physiques des crises. Parfois, l’utilisation d’un équipement de protection (par exemple,, rester dans un fauteuil roulant pour se protéger contre les blessures des attaques de chute) peut lui-même avoir un impact sur la mobilité des patients, alimentant un cercle vicieux qui limite leur capacité à être physiquement actif. De plus, le développement précoce de la dysphagie est fortement associé à un mauvais pronostic de crise à long terme (103) et affecte la capacité des patients à manger et à prendre des médicaments.
Les Patients atteints de LGS souffrent d’une perturbation du cycle du sommeil en raison de l’apparition de convulsions la nuit., La privation et/ou la perturbation du sommeil affectent les mécanismes neurophysiologiques et neurochimiques importants pour le processus d’apprentissage de la mémoire (104) et peuvent également entraîner un large éventail de troubles comportementaux, cognitifs et de l’Humeur, y compris l’hyperactivité, la réduction des notes scolaires et la dépression (105).
la prise en charge soigneuse des comorbidités chez les LG est un aspect essentiel des soins. Certains Dea peuvent provoquer ou exacerber des comorbidités (p. ex.,, troubles cognitifs, dépression), et le choix du traitement par Dea doit donc tenir compte de cette possibilité; par exemple, les benzodiazépines utilisées pour traiter les troubles du sommeil peuvent précipiter des crises toniques dans les LGS (106). Une attention particulière doit également être accordée à la possibilité d’interactions médicamenteuses de DEA avec les médicaments utilisés pour le traitement des comorbidités.,
en raison du fardeau important des convulsions et des comorbidités, et des effets secondaires des médicaments associés, la QV des patients atteints de SGL est altérée à de nombreux niveaux (physique, mental, social) tout au long de leur vie (3). L’impact physique du SGS et les mesures préventives prises pour minimiser cet impact affectent gravement la capacité des patients à participer aux activités quotidiennes, et la fréquentation scolaire est souvent perturbée. Les problèmes cognitifs et comportementaux nécessitent souvent des besoins spécifiques en matière d’éducation et de soins qui empêcheront la fréquentation scolaire ordinaire (3)., De plus, l’impact susmentionné des crises nocturnes sur le sommeil peut directement altérer la QV des patients (107). Les effets de LGS sur l’indépendance, la capacité de travail, la participation sociale et les relations personnelles continuent de nuire gravement à la QV des patients à l’âge adulte (3).,l’incidence du jor sur la qualité de vie des parents/aidants et des familles en raison de la restriction de la vie sociale et des problèmes relationnels entre les partenaires et les autres membres de la famille; le sentiment d’isolement, qui peut mener à la dépression; les problèmes de garde d’enfants, qui peuvent restreindre davantage la participation sociale et les possibilités de répit; l’épuisement physique et/ou la perturbation du sommeil; l’anxiété quant au moment où les crises se produiront, les perspectives d’avenir de la personne atteinte de LG et la stigmatisation sociale associée à la maladie; ainsi que les difficultés financières résultant du renoncement au développement de carrière en faveur de la prestation de soins (108).,
étant donné que les SSL constituent un fardeau majeur pour les patients et leurs soignants / familles, une approche multidisciplinaire et individualisée des soins est nécessaire, qui répond aux besoins médicaux, éducatifs, psychologiques et sociaux de chaque patient tout au long de sa vie (109). Idéalement, les besoins du patient et de son soignant/de sa famille devraient être réévalués chaque année, en tenant compte de facteurs tels que la santé physique du patient, son besoin potentiel de placement en institution (particulièrement à l’âge adulte) et le soutien apporté au soignant/à sa famille.,
domaines de recherche futurs
à l’heure actuelle, les DEA sont des médicaments anti-épileptiques et traitent donc les symptômes de l’épilepsie plutôt que sa(ses) cause (s). Les recherches futures devraient se concentrer sur l’élucidation de l’histoire naturelle de la LGS et sur la question de savoir si un traitement approprié peut avoir un impact bénéfique sur l’évolution de la maladie. Cela devrait inclure non seulement les effets du traitement pharmacologique, mais aussi le rôle des procédures chirurgicales telles que la callosotomie., Il a été suggéré que le contrôle précoce des convulsions chez LGS était associé à une amélioration des résultats neurocognitifs (2), mais la pertinence de ce contrôle dans Lgs de novo, ainsi que l’impact de la suppression des convulsions chez les enfants plus âgés et les adultes, est moins certaine et mérite une étude plus approfondie. Bien que l’on s’intéresse actuellement aux effets néfastes potentiels des DEA sur la cognition, des recherches sont nécessaires pour déterminer si d’autres traitements non liés aux DEA peuvent protéger contre les troubles cognitifs et/ou améliorer la cognition chez les patients atteints de SGL., En outre, la recherche est nécessaire pour développer des outils standardisés pour les soignants afin de mesurer régulièrement les changements dans les performances cognitives et le comportement des patients au fil du temps. L’élucidation quant à savoir s’il existe des génotypes spécifiques ou des mutations génétiques qui déterminent une susceptibilité au LGS est également nécessaire, ce qui pourrait orienter le développement d’outils de diagnostic.
compte tenu des problèmes associés au comptage des crises dans LGS (en raison de la fréquence des crises de goutte, de la durée des crises d’absence, etc.,), la valeur prédictive de l’utilisation d’autres paramètres dans la recherche clinique devrait être évaluée, comme le nombre de jours sans crise, plutôt que la fréquence des crises en soi. La caractérisation des facteurs qui prédisent la réponse au traitement chez les patients atteints de SGL serait également utile dans les milieux de recherche et de pratique clinique. D’autres domaines de recherche potentiels incluent la pertinence de la thérapie stéroïdienne dans LGS, et si elle peut améliorer la cognition , et les effets à long terme du traitement au cannabidiol dans LGS, en termes d’EI et de développement potentiel de tolérance.,
contributions des auteurs
tous les auteurs (JC, SA, MF, PS et AA) ont apporté une contribution substantielle à la conception de cet article ainsi qu’à l’analyse et à l’interprétation des données qu’il contient; ont participé à la rédaction de l’article ou à sa révision critique en raison de son contenu intellectuel important; ont donné leur approbation finale à la version à publier; et ont accepté d’être responsables de tous les aspects du travail en veillant à ce que les questions liées à l’exactitude ou à l’intégrité de toute partie du travail soient examinées et résolues de manière appropriée.,
déclaration de conflit d’intérêts
JC a siégé à des conseils consultatifs pour Eisai, UCB, Shire, Zogenix, Vitaflo et Nutricia, pour lesquels des paiements et des honoraires ont été versés à son département. Elle a participé en tant que chercheuse pour Vitaflo, GW Pharmaceuticals et Zogenix, avec des fonds versés à son département. Elle a également reçu des fonds de recherche de Vitaflo. Elle est soutenue par le National Institute for Health Research Biomedical Research Centre au Great Ormond Street Hospital for Children NHS Foundation Trust et University College London., SA a siégé à des conseils consultatifs pour Eisai, GSK, Novartis, Nutricia, Shire, Ultragenyx et Zogenix. Il a participé en tant que chercheur pour Advicenne Pharma, Eisai, UCB, Ultragenyx et Zogenix, avec des fonds versés à son hôpital ou à son laboratoire de recherche. MF a participé en tant qu’investigateur pour UCB, GW Pharmaceuticals, Pfizer et LivaNova, avec des fonds versés à son département. Elle a reçu des fonds de recherche de Eisai, UCB, LivaNova et Esteve, avec des fonds versés à son département. Elle a reçu des honoraires de conférencière de Eisai, UCB, LivaNova, Bial et Esteve., PS a reçu des honoraires de FB Health, Kolfarma s. r. l., UCB Pharma et Eisai Inc., et le soutien à la recherche du Ministère italien de la santé et de la Fondation Téléthon. AA a reçu des subventions de recherche de la Commission européenne, UCB Pharma et CaixaBank, avec des fonds versés à son département, et des honoraires de conseil D’Eisai, GW, Shire, Takeda, Zogenix et UCB Pharma.
Remerciements
Le soutien éditorial pour la préparation de ce manuscrit a été fourni par John Scopes de MXM Medical Communications.,
Financement
La Réunion du groupe de consensus pour l’élaboration de cette publication a été organisée et financée par Eisai Ltd. Le soutien éditorial pour la préparation de cette publication a été financé par Eisai Ltd. Les opinions exprimées dans cet article sont les opinions consensuelles indépendantes des auteurs et n’ont pas été influencées par le parrainage de tiers.
les Abréviations
Notes de bas de page
2. Arzimanoglou A, Resnick T. tous les enfants qui souffrent de chutes épileptiques n’ont pas nécessairement le syndrome de Lennox-Gastaut but mais beaucoup le font., Epileptic Disord (2011) 13(Suppl 1):S3–13. doi:10.1684/epd.2011.0422
CrossRef Full Text | Google Scholar
3. Kerr M, Kluger G, Philip S. Evolution and management of Lennox-Gastaut syndrome through adolescence and into adulthood: are seizures always the primary issue? Epileptic Disord (2011) 13(Suppl 1):S15–26. doi:10.1684/epd.2011.0409
PubMed Abstract | CrossRef Full Text | Google Scholar
5., Goldsmith IL, Zupanc ML, Buchhalter JR. Long-term seizure outcome in 74 patients with Lennox-Gastaut syndrome: effects of incorporating MRI head imaging in defining the cryptogenic subgroup. Epilepsia (2000) 41(4):395–9. doi:10.1111/j.1528-1157.2000.tb00179.x
PubMed Abstract | CrossRef Full Text | Google Scholar
8. International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes., Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia (1989) 30(4):389–99. doi:10.1111/j.1528-1157.1989.tb05316.x
CrossRef Full Text | Google Scholar
10. van Rijckevorsel K. Treatment of Lennox-Gastaut syndrome: overview and recent findings. Neuropsychiatr Dis Treat (2008) 4(6):1001–19. doi:10.2147/NDT.,S1668
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Gastaut H, Dravet C, Loubier D. Evolution clinique et prognostic du syndrome de Lennox-Gastaut. In: Lugaresi E, Pazzaglia P, Tassinari CA, editors. Evolution and Prognosis of Epilepsies. Bologna, Italy: Aulo Gaggi (1973). p. 133–54.
Google Scholar
17. EuroEPINOMICS-RES Consortium, Epilepsy Phenome/Genome Project, Epi4K Consortium., De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet (2014) 95(4):360–70. doi:10.1016/j.ajhg.2014.08.013
CrossRef Full Text | Google Scholar
18. Epilepsy Phenome/Genome Project Epi4K Consortium. Copy number variant analysis from exome data in 349 patients with epileptic encephalopathy. Ann Neurol (2015) 78(2):323–8. doi:10.1002/ana.,24457
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Larsen J, Johannesen KM, Ek J, Tang S, Marini C, Blichfeldt S, et al. The role of SLC2A1 mutations in myoclonic astatic epilepsy and absence epilepsy, and the estimated frequency of GLUT1 deficiency syndrome. Epilepsia (2015) 56(12):e203–8. doi:10.1111/epi.13222
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Hughes JR, Patil VK., Long-term electro-clinical changes in the Lennox-Gastaut syndrome before, during, and after the slow spike-wave pattern. Clin Electroencephalogr (2002) 33(1):1–7. doi:10.1177/155005940203300103
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Doose H. Myoclonic-astatic epilepsy. Epilepsy Res Suppl (1992) 6:163–8.
PubMed Abstract | Google Scholar
26. Patry G, Lyagoubi S, Tassinari CA., Subclinical « electrical status epilepticus” induced by sleep in children. A clinical and electroencephalographic study of six cases. Arch Neurol (1971) 24(3):242–52. doi:10.1001/archneur.1971.00480330070006
CrossRef Full Text | Google Scholar
28. King DW, Smith JR. Supplementary sensorimotor area epilepsy in adults. Adv Neurol (1996) 70:285–91.
PubMed Abstract | Google Scholar
29., Khan SA, Carney PW, Archer JS. Brève posture tonique asymétrique avec activité rapide diffuse à basse tension dans les crises provenant de la région pariétale mésiale. Épilepsie Res (2014) 108(10):1950-4. doi: 10.1016 / j. eplepsyres.2014.09.011
PubMed Abstract | CrossRef Texte Intégral | Google Scholar
33. Ohtahara S. syndrome de Lennox-Gastaut. Considérations dans son concept et sa catégorisation. JPN J Psychiatry Neurol (1988) 42(3):535-42.,
PubMed Abstract | Google Scholar
35. Pujar S, Calvert S, Cortina-Borja M, Chin RF, Smith RA, Cross JH, et al. Statistical process control (SPC) – a simple objective method for monitoring seizure frequency and evaluating effectiveness of drug interventions in refractory childhood epilepsy. Epilepsy Res (2010) 91(2–3):205–13. doi:10.1016/j.eplepsyres.2010.07.013
CrossRef Full Text | Google Scholar
39., Morrow J, Russell A, Guthrie E, Parsons L, Robertson I, Waddell R, et al. Malformation risks of antiepileptic drugs in pregnancy: a prospective study from the UK Epilepsy and Pregnancy Register. J Neurol Neurosurg Psychiatry (2006) 77(2):193–8. doi:10.1136/jnnp.2005.074203
PubMed Abstract | CrossRef Full Text | Google Scholar
41. Motte J, Trevathan E, Arvidsson JF, Barrera MN, Mullens EL, Manasco P, et al. Lamotrigine for generalized seizures associated with the Lennox-Gastaut syndrome., N Engl J Med (1997) 337(25):1807–12. doi:10.1056/NEJM199712183372504
CrossRef Full Text | Google Scholar
46. The Felbamate Study Group in Lennox-Gastaut Syndrome. Efficacy of felbamate in childhood epileptic encephalopathy (Lennox-Gastaut syndrome). N Engl J Med (1993) 328(1):29–33. doi:10.1056/NEJM199301073280105
CrossRef Full Text | Google Scholar
52., Ohtsuka Y, Yoshinaga H, Shirasaka Y, Takayama R, Takano H, Iyoda K. sécurité à Long terme et résultats des crises chez les patients Japonais atteints du syndrome de Lennox-Gastaut recevant un traitement adjuvant au rufinamide: une étude ouverte à la suite d’un essai clinique randomisé. Epilepsie Res (2016) 121:1-7. doi: 10.1016 / j. eplepsyres.2016.01.002
PubMed Abstract | CrossRef Texte Intégral | Google Scholar
55. Conry JA, Ng YT, Kernitsky L, Mitchell WG, Veidemanis R, Drummond R, et coll., Des dosages stables de clobazam pour le syndrome de Lennox-Gastaut sont associés à des améliorations soutenues de baisse-saisie et de Total-saisie sur 3 ans. Épilepsie (2014) 55(4):558-67. doi: 10.1111 / epi.12561
CrossRef Texte Intégral | Google Scholar
56. Munn R, Camfield P, Camfield C, Dooley J. Clobazam pour les troubles épileptiques réfractaires de l’enfance – un médicament supplémentaire précieux. Can J Neurol Sci (1988) 15(4):406-8.,
Google Scholar
60. Hussain SA, Zhou R, Jacobson C, Weng J, Cheng E, Lay J, et al. Perceived efficacy of cannabidiol-enriched cannabis extracts for treatment of pediatric epilepsy: a potential role for infantile spasms and Lennox-Gastaut syndrome. Epilepsy Behav (2015) 47:138–41. doi:10.1016/j.yebeh.2015.04.009
PubMed Abstract | CrossRef Full Text | Google Scholar
62., Geffrey AL, Pollack SF, Bruno PL, Thiele EA. Interaction médicamenteuse entre le clobazam et le cannabidiol chez les enfants atteints d’épilepsie réfractaire. Épilepsie (2015) 56(8):1246-51. doi: 10.1111 / epi.13060
PubMed Abstract | CrossRef Texte Intégral | Google Scholar
65. Biró A, Stephani U, Tarallo T, Bast t, Schlachter K, Fleger M, et coll. Efficacité et tolérabilité du pérampanel chez les enfants et les adolescents atteints d’épilepsies réfractaires: premières expériences. Neuropédiatrie (2015) 46(2):110-6. doi: 10.,1055/s-0035-1546276
PubMed Abstract | CrossRef Texte Intégral | Google Scholar
69. Je, Terao NN, Ng YT, Reisig W, Rubenstein JE, Kossoff EH. Efficacité du régime cétogène dans le syndrome de Lennox-Gastaut: examen rétrospectif de l’expérience d’une institution et résumé de la littérature. Dev Med Enfant Neurol (2012) 54(5):464-8. doi: 10.1111 / j. 1469-8749.2012. 04233.,x
PubMed Abstract | CrossRef Full Text | Google Scholar
72. Sharma S, Jain P, Gulati S, Sankhyan N, Agarwala A. Use of the modified Atkins diet in Lennox Gastaut syndrome. J Child Neurol (2015) 30(5):576–9. doi:10.1177/0883073814527162
PubMed Abstract | CrossRef Full Text | Google Scholar
76. Chambers A, Bowen JM. Electrical stimulation for drug-resistant epilepsy: an evidence-based analysis., En Ontario Health Technol Assess Ser (2013) 13(18):1-37.
PubMed Abstract | Google Scholar
77. Morris GL III, Gloss D, Buchhalter J, Mack KJ, Nickels K, Harden C. mise à jour des lignes directrices fondées sur des preuves: stimulation du nerf vague pour le traitement de l’épilepsie: rapport du Sous-Comité de développement des lignes directrices de L’Académie Américaine de Neurologie. Neurologie (2013) 81(16):1453-9. doi: 10.1212 / WNL.,0b013e3182a393d1
CrossRef Full Text | Google Scholar
78. Karceski S. Vagus nerve stimulation and Lennox-Gastaut syndrome: a review of the literature and data from the VNS patient registry. CNS Spectr (2001) 6(9):766–70. doi:10.1017/S1092852900001516
PubMed Abstract | CrossRef Full Text | Google Scholar
84. Rolston JD, Englot DJ, Wang DD, Garcia PA, Chang EF., Corpus callosotomy versus vagus nerve stimulation for atonic seizures and drop attacks: a systematic review. Epilepsy Behav (2015) 51:13–7. doi:10.1016/j.yebeh.2015.06.001
PubMed Abstract | CrossRef Full Text | Google Scholar
86. Cukiert A, Cukiert CM, Burattini JA, Lima AM, Forster CR, Baise C, et al. Long-term outcome after callosotomy or vagus nerve stimulation in consecutive prospective cohorts of children with Lennox-Gastaut or Lennox-like syndrome and non-specific MRI findings., Saisie (2013) 22(5):396-400. doi:10.1016/j.la saisie.2013.02.009
PubMed Abstract | CrossRef Texte Intégral | Google Scholar
87. Kwan SY, Lin JH, Wong TT, Chang KP, Yiu CH. Une comparaison des résultats de saisie après callosotomie chez les patients atteints de syndrome de Lennox-Gastaut et un effet positif ou négatif de l’histoire pour le syndrome de West. Saisie (2006) 15(7):552-7. doi:10.1016/j.la saisie.2006.06.,008
PubMed Abstract | CrossRef Full Text | Google Scholar
89. Katagiri M, Iida K, Kagawa K, Hashizume A, Ishikawa N, Hanaya R, et al. Combined surgical intervention with vagus nerve stimulation following corpus callosotomy in patients with Lennox-Gastaut syndrome. Acta Neurochir (Wien) (2016) 158(5):1005–12. doi:10.1007/s00701-016-2765-9
CrossRef Full Text | Google Scholar
93. Akman CI., Nonconvulsive status epilepticus and continuous spike and slow wave of sleep in children. Semin Pediatr Neurol (2010) 17(3):155–62. doi:10.1016/j.spen.2010.06.009
PubMed Abstract | CrossRef Full Text | Google Scholar
94. Walker M, Cross H, Smith S, Young C, Aicardi J, Appleton R, et al. Nonconvulsive status epilepticus: Epilepsy Research Foundation workshop reports. Epileptic Disord (2005) 7(3):253–96.,
PubMed Abstract | Google Scholar
98. Jurasek L, Ray L, Quigley D. Development and implementation of an adolescent epilepsy transition clinic. J Neurosci Nurs (2010) 42(4):181–9. doi:10.1097/JNN.0b013e3181e26be6
PubMed Abstract | CrossRef Full Text | Google Scholar
103. Ogawa K, Kanemoto K, Ishii Y, Koyama M, Shirasaka Y, Kawasaki J, et al., Étude de suivi à Long terme du syndrome de Lennox-Gastaut chez les patients présentant une déficience motrice et intellectuelle sévère: avec une référence particulière au problème de la dysphagie. Saisie (2001) 10(3):197-202. doi: 10.1053 / seiz.2000.0483
CrossRef Texte Intégral | Google Scholar
105. O’Brien LM. Les effets neurocognitifs de la perturbation du sommeil chez les enfants et les adolescents. Enfant Adolesc Psychiatre Clin N Am (2009) 18(4):813-23. doi:10.1016/j.chc.2009.04.,008
PubMed Abstract | CrossRef Full Text | Google Scholar
109. Arzimanoglou A, Resnick T. Diagnosing and treating epileptic drop attacks, atypical absences and episodes of nonconvulsive status epilepticus. Epileptic Disord (2011) 13(Suppl 1):S1–2. doi:10.1684/epd.2011.0408
CrossRef Full Text | Google Scholar
110. Kivity S, Lerman P, Ariel R, Danziger Y, Mimouni M, Shinnar S., Résultats cognitifs à Long terme d’une cohorte d’enfants atteints de spasmes infantiles cryptogéniques traités avec une hormone adrénocorticotrope à forte dose. Épilepsie (2004) 45(3):255-62. doi: 10.1111 / j. 0013-9580. 2004.30503.x
PubMed Abstract | CrossRef Texte Intégral | Google Scholar