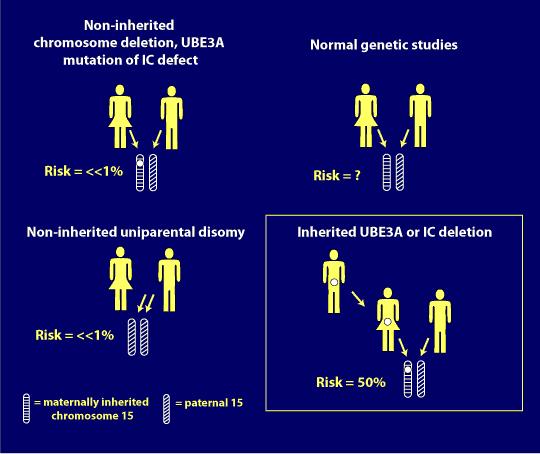
Schwere der Genotypen
Leider ist der häufigste AS-Genotyp, der positiv ist, in Bezug auf Symptome oder Merkmale tendenziell am schwersten.
Unten ist eine Illustration, die Dr. Charles Williams in seiner genetischen Präsentation auf der 2014 Canadian Angelman Syndrome Organization (CASS) Conference verwendete, um die Beziehung zwischen AS-Genotypen und der Schwere einiger AS-Merkmale zu zeigen.,
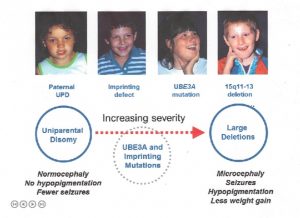
Rezidivrisiko beim Angelman-Syndrom
Ein genetischer Berater kann Sie über die Möglichkeit informieren, dass das Angelman-Syndrom durch Sammeln von Familienanamnese und Blutuntersuchungen auftritt oder erneut auftritt. Die folgenden Informationen können hilfreich sein, um das genetische Risiko des Angelman-Syndroms zu verstehen, sollen jedoch nicht die genetische Beratung ersetzen.
1., Gemeinsame Chromosomenlöschung:
Mehr als 98% der Chromosomenlöschungsinstanzen treten durch ein spontanes Ereignis auf und werden daher nicht vererbt; Das Rezidivrisiko ist <<1% für diese Familien. 1-2% der Deletionen treten jedoch aufgrund einer vererbten Anomalie im mütterlichen Chromosom 15 auf, z. B. einer ausgeglichenen Chromosomentranlokation. Eine weitere sehr kleine Gruppe (z.,, nur wenige Fälle in der Literatur berichtet), kann aufgrund einer sehr kleinen, mütterlich vererbten Chromosomeneletion, die einen kleinen Bereich um und einschließlich des UBE3A-Gens umfasst. In diesen Fällen ist das mütterliche Rezidivrisiko abhängig von der Art der vorhandenen Anomalie erhöht. Chromosomenstudie der Mutter, einschließlich FISCH, hilft, vererbte Chromosomenanomalien auszuschließen.
2. Väterliche uniparentale Disomie (patUPD):
Mehr als 99% der patUPD-Fälle treten als offensichtliches spontanes, nicht vererbtes Ereignis auf., Wenn eine Person AS aufgrund von patUPD hat und einen normalen Karyotyp hat, sollte dennoch eine Chromosomenanalyse der Mutter angeboten werden, um die seltene Möglichkeit auszuschließen, dass eine robertsonianische Translokation oder ein Markerchromosom ein prädisponierender Faktor war (z. B. über die Generation von mütterlichem Gameten, der für Chromosom 15 nullisomisch war, mit anschließender postzygotischer „Korrektur“ zur väterlichen Disomie).
3. Imprinting Center (IC) Defekt:
Es gibt zwei Arten von IC-Defekten: Löschungen und Nicht Löschungen., Nicht löschende Ereignisse scheinen nicht vererbt zu sein und haben ein <1% Rezidivrisiko. Die meisten Deletionen werden nicht vererbt, aber ein erheblicher Teil von ihnen ist (dh mütterlich vererbt), und diese bergen ein 50% iges Risiko für ein Wiederauftreten.
4. UBE3A-Mutationen:
Die UBE3A-Mutation kann entweder spontan auftreten (z. B. nicht vererbt und ohne erhöhtes Rezidivrisiko) oder mütterlicherseits vererbt werden und ein Rezidivrisiko von 50% aufweisen (siehe unten zur Prägung der Vererbung).
5., Personen ohne bekannten Mechanismus (alle 4 oben genannten Mechanismen wurden eliminiert):
Für Eltern von AS-Personen mit offensichtlichen normalen Gentests (keine Hinweise auf Deletion, Prägedefekt, UPD-oder UBE3A-Mutation) und damit ihre Kinder werden nur klinisch diagnostiziert, es ist nicht bekannt, was das Rezidivrisiko ist. Ein erhöhtes Risiko scheint wahrscheinlich, überschreitet aber wahrscheinlich nicht 10%.
6. Keimzellmosaizismus:
Dieser Begriff bezieht sich auf ein Phänomen, bei dem ein genetischer Defekt in den Zellen der Gonade (Eierstock im Fall der Mutter) vorhanden ist, nicht jedoch in anderen Körperzellen., Dieses Auftreten kann zu Fehlern bei der Risikobewertung führen, da ein Gentest, beispielsweise an den Blutzellen einer Mutter, normal ist, wenn tatsächlich ein genetischer Defekt in den Keimzellen ihres Eierstocks vorhanden ist. Glücklicherweise tritt der Keimzellmosaizismus sehr selten auf. Nichtsdestotrotz wurde es in AS beobachtet, das durch die Mechanismen der großen Chromosomen Deletion, der Prägung der Center Deletion und der UBE3A Mutation verursacht wurde.
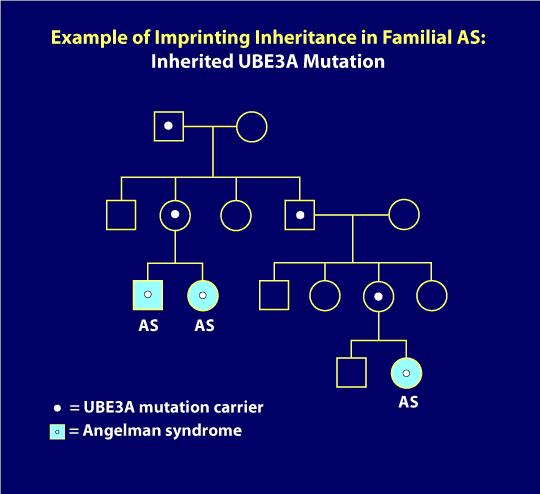
7., Prägung Vererbung:
UBE3A Mutationen und Prägung Zentrum Deletionen können Prägung Vererbung zeigen, wobei ein Träger Vater auf dem genetischen Defekt an seine Kinder weitergeben kann, ohne dass es irgendwelche Probleme verursacht, aber wenn eine Frau gibt diesen gleichen genetischen Defekt auf ihre Kinder, unabhängig vom Geschlecht ihres Kindes, wird dieses Kind ALS haben. Das Stammbaumdiagramm unten veranschaulicht die Prägung der Vererbung. Hier, WIE erst aufgetreten ist, nachdem eine Trägermutter den Gendefekt weitergegeben hat (zum Beispiel wie bei den beiden Geschwistern mit WIE auf dem linken unteren Teil des Stammbaums abgebildet)., Darüber hinaus hat ein entfernter Cousin in dieser Familie auch aufgrund des prägenden Erbes. In dem Diagramm haben Individuen mit den hellblauen Kreisen oder Quadraten, wie aber alle anderen in der Familie klinisch normal ist. Die weißen Punkte stellen asymptomatische, normale Träger der AS-Mutation dar. Wenn festgestellt wird, dass ein AS-genetischer Mechanismus vererbt wird, können Gentests von Familienmitgliedern normalerweise Träger des Gendefekts identifizieren. Wie Sie sich vorstellen können, wird in diesen Situationen eine professionelle genetische Beratung empfohlen.